


Keywords
Abstract
NMR spectroscopy of paramagnetic metal complexes was long considered a challenging area of research due to signal broadening and difficulties in spectral interpretation. However, modern theoretical and experimental developments have transformed this physicochemical method into an indispensable tool, particularly for the design of functional molecular materials. This review offers the first comprehensive view of paramagnetic NMR spectroscopy, combining a detailed analysis of the physical foundations and experimental techniques with a summary of modern theories and approaches. Particular attention is given to the method’s potential for establishing the structure, electronic structure, and magnetic properties of complexes, determining the parameters of magnetic interactions critical for the creation of single-molecule magnets, spin switches, and other precursors to future functional materials. The synergistic effect of combining NMR spectroscopy with magnetometry and EPR is emphasized, allowing one to overcome the problem of overparameterization and obtain reliable data on the electronic structure of the complex.
The bibliography includes 157 references.
1. Introduction
The first studies in nuclear magnetic resonance (NMR) spectroscopy date back to the 1940s.[1][2] Initially, this method was used only for studying diamagnetic compounds, as paramagnetic compounds presented significant challenges due to extremely short nuclear relaxation times and significant chemical shifts. In the 1960s, the first attempts to apply NMR spectroscopy to paramagnetic compounds were made.[3] However, due to the difficulty of interpreting the resulting data, such studies were sporadic.
The foundation for understanding hyperfine interactions in paramagnetic NMR was laid by the development of the theory of electron paramagnetic resonance (EPR), the basic principles of which were developed by the Soviet physicist E.K.Zavoisky* in 1944. The 1950s and 1960s saw significant advances in NMR spectroscopy of paramagnetic compounds. New theories and data analysis methods were developed that provided more accurate information on the structure and dynamics of paramagnetic compounds,[4-6] such as McConnell’s contact shift theory,[7] McGarvey’s paramagnetic shift theory,[8] and Bleaney’s theory.[9]
Modern NMR spectroscopy of paramagnetic compounds is an effective tool for studying the structure, magnetic properties, and chemical bonding of molecules. It finds application in many fields, including chemistry, physics, biology, and medicine. For example, the concept of paramagnetic labeling is used to study the structure of biological macromolecules. Selective labeling of the macromolecule under study allows for the recording of a three-dimensional structure comparable in quality to X-ray diffraction methods. The principles of paramagnetic NMR spectroscopy (pNMR) can also be transferred to magnetic resonance imaging (MRI), which is based on similar physical principles. For example, the use of molecular paramagnetic labels in vivo allows for the influence of MRI contrast by accelerating the relaxation of water molecules exchanging between the coordination sphere of the paramagnetic center (inner sphere) and the bulk solvent (outer sphere). One area of active research in the pNMR technique is the development of new markers and contrast agents for medical imaging. This opens the possibility of using NMR spectroscopy to visualize biomolecules in vivo, which is important for the diagnostics and treatment of various diseases.
Due to the constant development of technology and data analysis methods, NMR spectroscopy of paramagnetic compounds continues to evolve and find new applications in science. In particular, it is used to conduct detailed studies of the structure, electronic structure, and magnetic properties of new paramagnetic metal complexes, which are precursors to promising functional materials such as quantum computers, spintronic devices, and high-density data storage.[10] Special attention should be paid to the growing number of studies combining the analysis of data from traditional physicochemical methods in a magnetic field (magnetometry and EPR) with paramagnetic NMR spectroscopy.[11-16] These examples demonstrate the growing importance of NMR spectroscopy as a method for studying paramagnetic compounds.
This review presents the state-of-the-art of NMR spectroscopy of paramagnetic metal complexes and provides application examples. A unique, comprehensive analysis of the method is provided, including a discussion of its fundamental principles (the nature of hyperfine shifts, relaxation mechanisms, and partial orientation effects), as well as practical aspects (selection of spectral recording parameters, applicability limits of 2D techniques, and consideration of chemical exchange).
* See E.Zavoisky. J. Phys. USSR, 9, 211 (1945).
2. Theoretical basis of NMR spectroscopy of paramagnetic compounds
In diamagnetic molecules, all electrons are paired such that their ground state corresponds to a net spin moment of zero for the electron pair. Therefore, the influence of electrons on the magnetic field at local locations within the molecule is very limited, as evidenced by the observed chemical shifts of nuclei in diamagnetic compounds, parts per million (ppm). Paramagnetic molecules have one or more unpaired electrons, which possess a spin moment and, accordingly, acquire a magnetic dipole moment in a magnetic field,
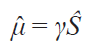
where m̂ and Ŝ are the quantum-mechanical operators of the magnetic dipole and spin moments, respectively, and γ is the gyromagnetic ratio. Since the gyromagnetic ratio of an electron is three to four orders of magnitude larger than that of nuclei, it is the properties of unpaired electrons that determine the magnetic properties of a molecule and, consequently, influence the characteristics observed in all physicochemical methods that employ an external magnetic field. With regard to NMR spectroscopy in solution, the most important effects caused by unpaired electrons are the following:
— the emergence of significant magnetic susceptibility in a solution of a paramagnetic compound ;
— the interaction of the nuclear magnetic moment with the electron magnetic moment (HFC);
— enhancement of nuclear magnetic relaxation due to the dynamics of the electron magnetic moment;
— anisotropic orientation of molecules in solution due to the spin dynamics of electrons;
— a temperature dependence of all observed effects due to changes in the population of electron energy levels.
Each of these effects is reflected in NMR spectra and can be used in appropriate experiments to study the magnetic properties of molecules and their electronic and nuclear structures. Fig. 1 shows a general diagram of the resulting effects and their relationship to magnetic coupling parameters.
![[{"id":"vzvtZiN94X","type":"paragraph","data":{"text":"Diagram of paramagnetic effects in NMR and their relationships to magnetic coupling parameters. The numbers of the corresponding mathematical expressions are given in parentheses."}}]](/storage/images/resized/9BhrdVw62tnIVstSv4HEd0FCSn5UWQfWiMJvn6Rl_xl.webp)
2.1. Change in magnetic susceptibility of the solution
Since a paramagnetic molecule has a significant magnetic dipole moment, a solution of such a compound will have a macroscopic magnetization M→ in a magnetic field of strength H→
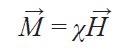
where c is the bulk magnetic susceptibility of the solution. Thus, in NMR experiments, the effective magnetic field in a solution is the sum of the external and induced fields.[17] Consequently, the change in the effective field ∆H will be proportional to the magnitude of the magnetization M
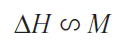
A change in the effective magnetic field leads to a change in the observed value of the chemical shift of any nucleus ∆δ present in the given solution, which is proportional to the bulk magnetic susceptibility of the solution
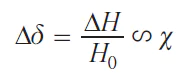
This equation leads to two important conclusions regarding pPMR. First, the change in the chemical shift of the nuclei of a paramagnetic molecule will occur not only due to direct interaction with the magnetic moment of electron, but also due to a change in the macroscopic magnetic susceptibility of the entire solution. Thus, not only the nuclei of the paramagnetic molecule in the solution, but also the nuclei of other molecules in the solution will undergo changes in chemical shift. Second, the observed ∆δ value can be used to determine the magnetic susceptibility of the solution. This approach, discovered in 1959, is called the Evans method[18] and is still widely used to study various paramagnetic molecules, such as single-molecule magnets and spin switches.[19]
The quantitative dependence of ∆δ and magnetic susceptibility depends on both the sample shape and the direction of the external magnetic field in the spectrometer. Since the vast majority of samples used for NMR spectroscopy are currently cylindrical, and the magnetic field vector of the superconducting magnet is aligned with the sample’s rotation axis, we present an expression specifically for this case (Eq. (5)):[20]
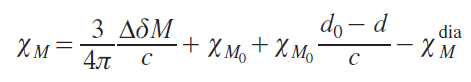
where χM is the molar magnetic susceptibility (cm3 mol–1, Δδ is a chemical shift change (ppm), M is the molar mass of a paramagnetic compound (g mol–1), c is the concentration of a paramagnetic solute (g cm–3), χM0 is the molar magnetic susceptibility in the absence of the paramagnetic compound (cm3 g–1), d and d0 are the densities of the solution of the paramagnetic compound and pure solvent, respectively (g cm–3), χMdia is the diamagnetic contribution to the molar magnetic susceptibility of a paramagnetic compound (cm3 mol–1). In this case, the CGS system units are traditionally used.
In practice, the term in Eq. (5) is neglected, since it is equal to zero at infinite dilution,[20] which leads to the simplified Eq. (6):
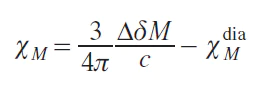
To estimate the diamagnetic component, an additive scheme of diamagnetic contributions of atoms and chemical bonds is used:[21]
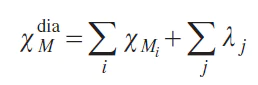
where χMi is the diamagnetic susceptibility of every i-th atom in the molecule, λj is the contribution made by each j-th chemical bond. These characteristics are for reference only.[21]
Since the temperature dependence of magnetic susceptibility is often measured, it should be noted that the concentration c in Eq. (6) will change depending on the temperature (T ) due to the change in the density of the solution:
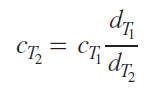
where d is the density of the solvent at the given temperature, indicated as a subscript; T1 and T2 are two temperatures.
Evans method is experimentally implemented using a coaxial insert in the NMR tube to simultaneously record the spectra of a paramagnetic solution and a pure solvent. Despite the simplicity and elegance of this method, the results obtained using it often have significant errors.[22][23] The reasons for this are as follows:
— difficulties in obtaining an absolutely pure substance for analysis;
— differences in densities of solutions with and without a paramagnetic compound;
— inability to accurately determine the diamagnetic correction;
— additional chemical shift caused by intermolecular interactions of molecules with the paramagnetic molecule;
— partial evaporation of the solvent when operating at elevated temperatures, causing a change in concentration.
Taking these sources of error into account, the error in determining magnetic susceptibility using the Evans method can be up to 10 – 15%.
2.2. Effects of partial orientation of molecules in a magnetic field
In general, the magnetic susceptibility χ is a symmetric second-rank tensor*
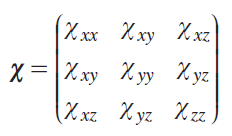
This means that this quantity can take on different effective values depending on the orientation of the object in the magnetic field, i.e., exhibit anisotropy. The molecular orientation that results in a higher magnetic susceptibility is more energetically favorable in the magnetic field and, consequently, more populated. Therefore, paramagnetic molecules in the solution will be oriented anisotropically. This phenomenon leads to deviations in some observed properties of the sample from isotropic values calculated based on the equiprobable orientation of the molecule.
From the pNMR point of view, the effect of partial orientation is reflected mainly in the appearance of residual dipolar and quadrupolar couplings (RDC and RQC). In most cases, this effect has an extremely weak impact on other NMR characteristics (chemical shift, relaxation, magnetic susceptibility of the solution, etc.), and therefore, as a rule, it is neglected.[22][23] Dipole interactions between nuclei average to zero in isotropic solutions,[24] so the splittings associated with them are almost never observed in the NMR spectra of diamagnetic compounds. For solutions of paramagnetic compounds exhibiting significant anisotropy of magnetic susceptibility, residual dipole interaction caused by partial orientation may be observed. In the spectra, this effect will manifest itself as a change in the multiplet splitting constant compared to the scalar spin-spin interaction constant of the diamagnetic analog by the following value:
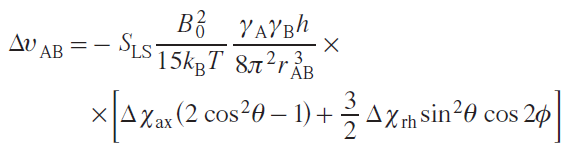
where SLS is the Lipari – Szabo order parameter depending on the intra-nuclear mobility of nuclei A and B (takes values from 0 to 1); B0 is the external magnetic field, kB is the Boltzmann constant; Т is temperature; h is the Planck constant; rAB is the distance between nuclei A and B; Δχax and Δχrh are axial and rhombic components of the magnetic susceptibility tensor; q and f are azimuthal and equatorial angles in the spherical coordinate system.[25] From the above equation it follows that the RDC effect is preferentially observed in high magnetic fields (>14 T) and for closely spaced nuclei, such as geminal protons (CH2) or the 13C – 1H pair of nuclei. The residual dipole interaction is observed even for equivalent nuclei for which the isotropic constant J does not appear in the spectra.[26][27]
For quadrupole nuclei (with nuclear spin I > 1/2) the magnitude of the residual quadrupole interaction is expressed as
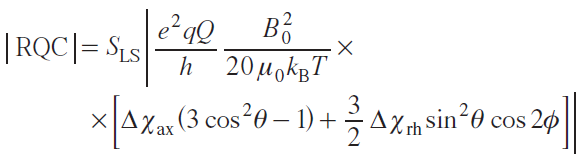
where
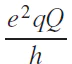
* Hereinafter, the tensor is indicated in bold.
2.3. Hyperfine shift
By definition, a chemical shift is the resonant frequency of a nucleus in a field of a given magnitude relative to some generally accepted standard. For a diamagnetic compound, the chemical shift ranges of nuclei in a given environment are generally well known. In the spectra of paramagnetic compounds, due to the presence of unpaired electron density, these rules are not applicable, since electrons have a magnetic moment and create an additional magnetic field at the location of a particular nucleus. Thus, the total magnetic field strength (H), in addition to the spectrometer field (H 0) and the field induced by diamagnetic electrons (H dia), has another additive term, which is responsible for the unpaired electrons (H eff)
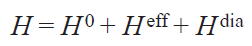
Within the spin Hamiltonian (SH) formalism, the summand H eff arises from the HFC between the electrons of the system and the observed nucleus in the NMR spectra. The static SH of a simple electron system with spin (S) equal to 1/2 in the absence of excited states,[29] which takes into account the interaction of the electron with the nucleus, is written as
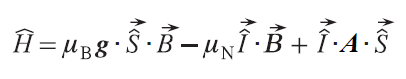
where g is an electronic g tensor, mB is the Bohr magneton, mN is the nuclear magneton, A is the HFC tensor,
Fig. 2 shows the energy diagram for the simplest system of two spins, S = 1/2, with an isotropic HFC constant (the field is directed along the Z axis). In such a system, two allowed nuclear spin transitions are possible, so the NMR spectrum should show two signals at the nuclear Zeeman interaction frequencies, one increased and one decreased by half the isotropic HFC constant. Since typical electron–proton HFC constants are several MHz, the chemical shift of such signals should be on the order of tens of thousands of ppm. This, however, does not occur, as these transitions are averaged out due to rapid electron relaxation. As a result, only one weighted signal is observed in the NMR spectrum. The position of this signal is shifted relative to the nuclear Zeeman interaction frequency due to the different populations of the Kramers doublets (KDs),
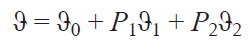
где ϑ is the observed resonant frequency, ϑ0 is the resonant frequency in the absence of unpaired electron effects, P1 and P2 are the populations of the first and second KDs, respectively, ϑ1 are ϑ2 are the resonant frequencies of the nuclei of these KDs.
![[{"id":"JjoG_dLXZZ","type":"paragraph","data":{"text":"Energy level splitting diagram for an electron (S = 1/2) interacting with a nucleus (I = 1/2). En is the energy of the n-th level, mI , mS are the magnetic quantum numbers of the nucleus and electron, respectively."}}]](/storage/images/resized/b0BYUZ8ACjHl4eNTBsjR4jhGc1XtAn8tiflBG6Zb_xl.webp)
In the case of systems with a large number of unpaired electrons (S > 1/2), having i Kramers doublets, Eq. (14) is:
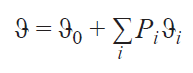
It should be noted that all subsequent formulas and models of chemical shift assume that a single electronic state is being considered, characterized by its unpaired electron density distribution and electron interactions. The most general theory of chemical shift at the moment, as described in monographs,[30][31] provides the following expression for the chemical shift observed in the NMR spectrum of a paramagnetic compound:
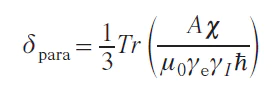
where χ is the magnetic susceptibility tensor, γe is the gyromagnetic ratio of the electron, γI is the gyromagnetic ratio of a nucleus, ħ is the reduced Planck constant (ħ = h/2π), m0 is the vacuum permeability.
The HFC tensor is usually divided into two components — isotropic and dipolar:
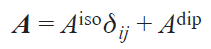
where Aiso is the trace of the HFC matrix, Adip is the traceless dipolar part of the HFC matrix, dij is the Kroneker delta, i, j = x, y, z.
Chemical shift is also usually divided into two terms: contact and dipolar (pseudocontact), while simple relationships and additivity are preserved:
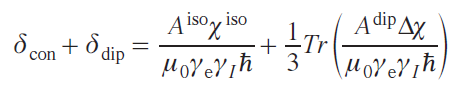
where Δχ is a traceless part of the magnetic susceptibility tensor matrix (dipolar), δcon is the contact shift, δdip is the dipolar shift.
Therefore, the total chemical shift in the spectrum of a compound includes the following three terms:
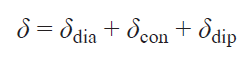
The above calculations are approximate; in reality, the theory of hyperfine shift is more complex, and has been significantly developed in recent years.[32-34] In particular, even for a system with S = 1/2, there are nine contributions to the hyperfine shift.[35] However, these additional contributions have little effect on the magnitude of the observed shift, so they have not yet been determined experimentally but only theoretically.[35][36] Moreover, other parameters, such as small structural changes in the solution, dynamic effects, and the influence of the solvent environment, can cause more significant changes in the hyperfine shift. Therefore, a simplified Eq. (19) is usually used to determine the hyperfine shift. The contact shift arises from an interaction that is proportional to the probability of finding an unpaired electron on the nucleus, i.e., the isotropic HFC constant,[8] which can be expressed through the spin density in the point of the nucleus position (ρN):
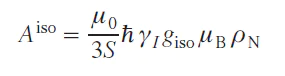
where giso is an isotropic value of the g-tensor. It is worth noting that Aiso in Eq. (20) is nonzero only in the case of an admixture of s orbitals in the electron wave function, since only they do not have a node at the position of the nucleus. This case can be realized through two mechanisms:
— delocalization of the spin density belonging to the atomic orbitals of the metal ion (or ligand, if the ligand is a radical) to molecular orbitals localized at the site of the nucleus being studied;
— spin polarization, the effect of which is that the spin density polarizes adjacent orbitals, allowing the contact shift to move along the bond chain.
However, as a rule, the contact shift becomes insignificant as the number of bonds between the unpaired electron localization center and the nucleus increases to 6 – 7.
For systems with S = 1/2 and in the absence of zero-field splitting (ZFS), the expression for the magnetic susceptibility tensor elements is as follows:
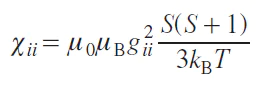
where i = x, y, z.
Thus, the value of the contact shift for systems with S = 1/2 and in the absence of ZFS is
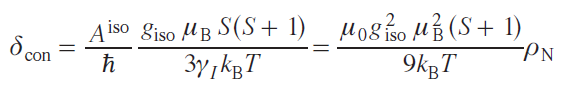
where ρN is the spin density at the position of a nucleus,
In the S > 1/2 systems, the ZFS effect can occur, in which the degeneracy in the magnetic quantum number of the spin projection (MS) is removed in the absence of an external magnetic field due to the residual spin – orbit interaction.[37] In this case, there is an approximate expression for estimating the contact shift:
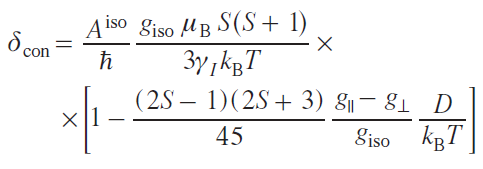
where g|| and g┴ are longitudinal and transverse components of the g-tensor, D is the zero-field splitting energy.
A more accurate approach to modeling the contact is to directly calculate the energy levels and resonant transitions between them[15] using the SH formalism
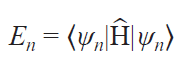
where ψn are SH eigenfunctions.
Various forms of spin-orbit interactions are described, which are chosen depending on the class of complexes being studied. For example, for complexes of metals such as nickel(II), manganese(III), titanium(III), and others, which exhibit weak magnetic anisotropy, the zero-field splitting formalism is used, within which the residual spin – orbit interaction is considered as a perturbation of the spin moment operator.[37]
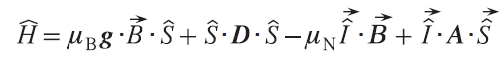
where g is electronic g-tensor, D is zero-field splitting tensor.
In the case of complexes exhibiting high magnetic anisotropy, a different model is needed that takes into account the spin – orbit interaction directly (Eq. (26)).[16] Such complexes include all single-molecule magnets by definition, as well as complexes of metal ions such as CoII, FeII, Fe III, etc.
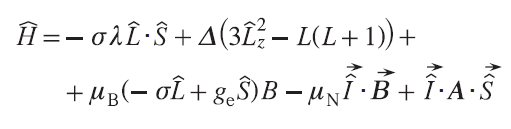
where L̂ is the orbital angular momentum operator of electron, λ is spin–orbital interaction energy, σ is spin – orbit coupling reduction factor, and Δ is the crystal field parameter. This expression is given for axially symmetric complexes, taking into account a single ligand field parameter. For more anisotropic ions, such as lanthanides, higher-order ligand field parameters must be considered.[38][39]
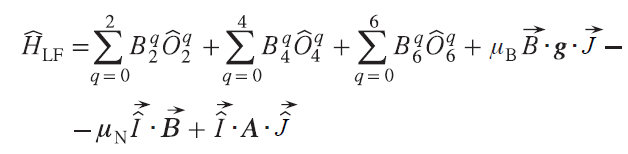
where Ôiq, i = 2, 4, 6 (Stevens operators) are polynomial functions of the total angular momentum operators Ĵz, Ĵ+, Ĵ–, tabulated in the work,[40] Biq are ligand field parameters (in the Stevens operator formalism).
The contact shift is calculated directly as the difference between the weighted average resonant frequency of a nucleus (in the approximation of fast dynamics) and the Larmor frequency at a given magnetic field strength
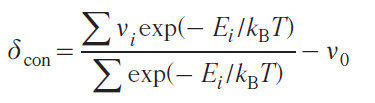
νi is the transition frequency of the i-th Kramers doublet, Ei is the energy of the i-th Kramers doublet, ν0 is the Larmor frequency of the nucleus in the absence of interaction with unpaired electrons.
A consequence of the dipole – dipole interaction between unpaired electrons and the nucleus is a pseudocontact shift.[41] Rapid molecular motion in solution averages out this interaction. However, due to the presence of magnetic anisotropy, averaging does not reach zero, leaving a nonzero chemical shift, referred to as the dipolar or pseudocontact shift. The anisotropy of the magnetic susceptibility tensor has axial and orthorhombic components

The components of the magnetic susceptibility tensor are also related to the energy levels, which are the eigenvalues of the above-mentioned SHs (25) – (27), as follows.[16] Magnetization is the partial derivative of energy with respect to the magnetic field
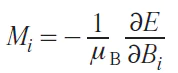
where Bi is the magnetic field at i = x, y, z.
Given the n-th number of energy levels of the electron spin state in a molecule, the magnetization, taking into account the populations of these levels in accordance with Boltzmann statistics, is expressed as follows:
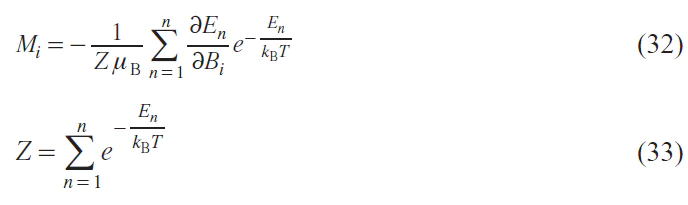
Alternatively, Eq. (32) can be simplified as follows:
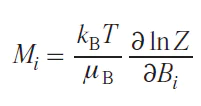
The components of the magnetic susceptibility tensor will be equal to
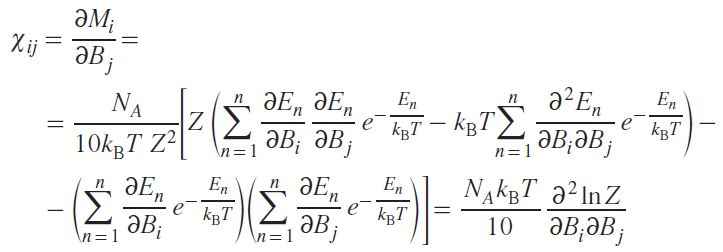
where NA is Avogadro’s number, i, j = x, y, z.
To fully describe the dipole shift, it is necessary to know the HFC tensor for each nucleus. In most cases, this is unknown, so various models use a number of assumptions. The simplest model (the point dipole model) assumes that the unpaired electron density is predominantly concentrated at the paramagnetic nucleus. This approximation does not work well in cases of large delocalization of the unpaired density, e.g., in organic radicals.[42] With this approach, the energy of the dipole–dipole interaction of two magnetic moments is determined as
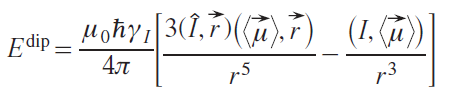
where ⟨μ→⟩ is the average magnetic moment of unpaired electrons, Î is the nuclear spin operator, r→ is the vector connecting the electron and the nucleus, γI is the gyromagnetic ratio of a nucleus.
The same expression can alternatively be represented, separating the spatial part from the magnetic characteristics, and also taking into account the direct proportionality between the magnetic moment of the electron and the magnetic susceptibility, so that the dipole chemical shift becomes equal to
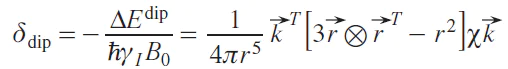
where k→ is the unit vector in the direction of the external magnetic field.
The expression in square brackets is the so-called symmetric dipole tensor. Due to the rapid rotational motion of the molecule compared to the NMR time scale, the observed quantity is the averaged shift. Spherical averaging gives the following expression, first obtained by McConnell.[7]
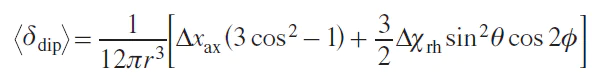
Using the expression for the components of the magnetic susceptibility tensor, an expression for the dipole shift in the case of axial symmetry can be obtained:
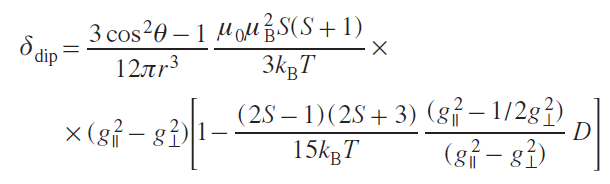
Despite all its advantages and usability in practical applications, the point interaction model has one undeniable drawback. It does not account for the interaction of the distributed density of the unpaired electron with the observed nuclei. A more general equation that takes this drawback into account was proposed[43] but has not yet found widespread use.
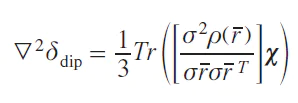
where ρ(r̅ ) is the electron density distribution function.
From Eq. (38) it follows that the dipole shift is easily predicted based on the molecular geometry and magnetic anisotropy. Geometry can be determined using an X-ray diffraction experiment on a single-crystal sample or quantum chemical optimization. Magnetic anisotropy can be determined in the following ways:
— magnetometric measurement of a single-crystal sample in various orientations;
— determination of the g-tensor anisotropy using EPR spectroscopy in the absence of ZFS effects;
— in the case of an isotropic g-tensor, using methods for measuring the ZFS energy (e.g., by terahertz EPR spectroscopy (THz-EPR));
— by measuring residual dipole and quadrupole interactions.
Thus, the chemical shift in the NMR spectrum of a paramagnetic compound depends on a number of factors, each of which can be investigated if the others are known along with the experimental shift value, namely:
— spin density distribution in a molecule (contact shift);
— molecular geometry (dipole shift);
— magnetic anisotropy (dipole shift).
Contact shift is rarely used to study paramagnetic molecules, so the challenge often arises of separating the dipole contribution from the others. The orbital contribution is typically estimated from the chemical shift of the closest diamagnetic compound. In the case of 3d-metal complexes, this is a complex with the same ligand and a related diamagnetic metal ion. If none is available, the free ligand is used.
The HFC constant can generally be measured using EPR spectroscopy; however, in practice, a molecule contains a large number of magnetoactive nuclei with similar constants. Therefore, the value of Aiso (or ρN) is typically estimated using quantum chemical calculations, which allows for the determination of the contact contribution. The separated dipole contribution can then be used in two ways: with a known geometry of the complex, it allows to determine the magnetic anisotropy, or with a known magnetic anisotropy, it allows to determine the geometry of the complex. These two approaches underlie two types of research: magnetic and structural. This review further examines a number of studies of magnetic properties.
2.4. Relaxation enhancement
The presence of HFC between the magnetic moments of the unpaired electron and the nucleus causes not only the hyperfine shift but also the enhancement of nuclear relaxation in the paramagnetic molecule. The HFC energy fluctuates randomly over time through three mechanisms:
1) molecular rotation, which changes the direction of the vector r→ in a magnetic field (see Eq. (36));
2) electron relaxation, which changes the effective value of magnetic anisotropy;
3) chemical exchange, which leads to a change in the coordinates of interacting particles.
Each mechanism is characterized by the correlation time (τ) of the corresponding process; this can be the correlation time (τr) of the rotational reorientation of the molecule, longitudinal and transverse electron relaxation times (τe1 and τe2), as well as the residence time τM) characteristic time corresponding to the chemical exchange processes.
Since the HFC consists of isotropic (contact) and dipole parts, the contributions of these interactions to the relaxation enhancement are also distinguished. Furthermore, there is another relaxation mechanism associated with the interaction of the magnetic moment of the nucleus with the average magnetic moment of the ensemble of surrounding molecules of the same name, which is called the Curie mechanism. Thus, the relaxation enhancement (Ri) includes three components
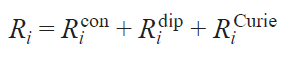
where i = 1 and 2 for longitudinal and transverse relaxations, respectively. The corresponding correlation times are added up similarly:
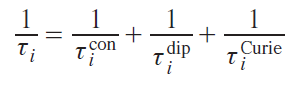
In turn, the correlation times of each mechanism consist of the correlation times of the processes that cause relaxation via that mechanism. For example, all three mechanisms (molecule reorientation, electron relaxation, and chemical exchange) lead to a change in the energy of the dipole – dipole interaction between the nucleus and the electron, i.e., they cause dipole relaxation, as follows from Eq. (36). Thus, the correlation time of this type of relaxation is
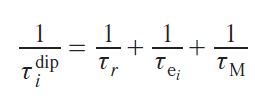
where τr, τei, τM are correlation times corresponding to the mechanisms of molecular reorientation, electron relaxation and chemical exchange.
In the case of contact relaxation, the reorientation of the molecule in a magnetic field does not affect the magnitude of the contact interaction, as it is associated exclusively with the intramolecular structure. Other mechanisms (electronic relaxation and chemical exchange) lead to fluctuations in the spin distribution in the molecule and therefore affect this type of relaxation
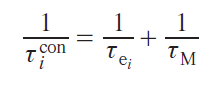
Curie relaxation is based on the interaction of the nucleus with electrons outside the molecule, so the relaxation of electrons within the molecule does not affect the correlation time of this type of relaxation. On the other hand, molecular motion and chemical exchange lead to a change in the orientation of the molecule relative to the environment
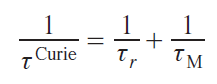
Dipole relaxation is typically described by the Solomon equations.[41] This approach considers only the Zeeman interaction. Other interactions, including anisotropic ones such as ZFS, are neglected, which significantly limits this approach for anisotropic metal ions. In particular, for the cobalt(II) ion in the high-spin state, magnetic anisotropy is almost entirely explained by ZFS, not by the Zeeman interaction. However, for many transition ions, such as MnII and CuII, the Solomon equations are applicable. Omitting the mathematical calculations, expressions for the enhancement of longitudinal and transverse relaxation can be written as follows:
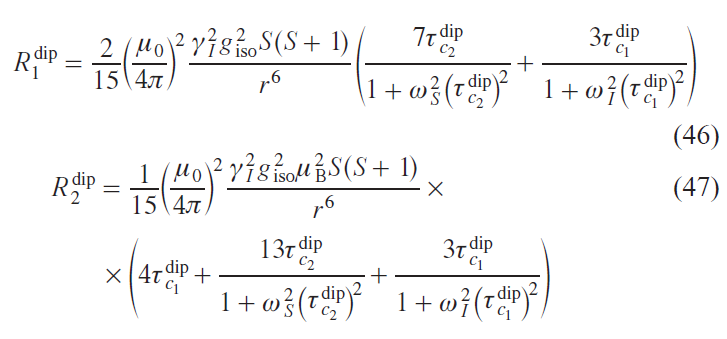
where r is the distance from the nucleus to the center of spin density localization, ωS and ωI are the Larmor frequencies of the electron and nucleus, respectively.
Within the limits of these approximations, expressions were obtained for the enhancement of contact relaxation and Curie relaxation:
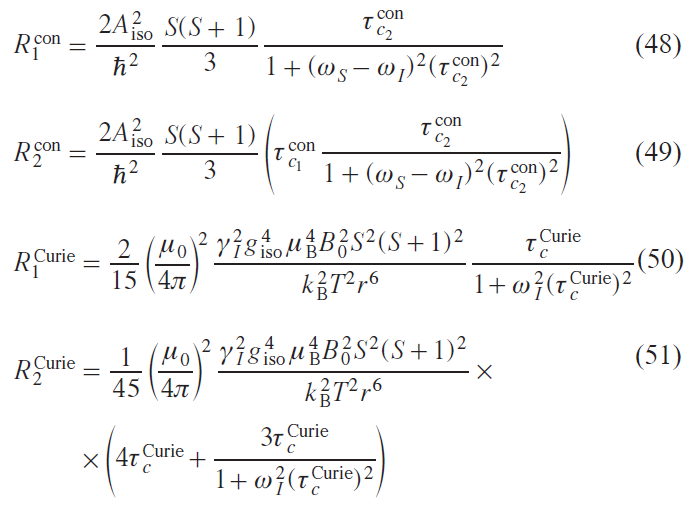
where Ricon is the contact contribution to relaxation, Ridip is the dipole contribution to relaxation, RiCurie is the contribution of the Curie mechanism to relaxation.
Noteworthy, the expressions given for the enhancement of relaxation by various mechanisms were derived taking into account the following approximations:
1) the unpaired electron is a point magnetic dipole (point-dipole approximation);
2) molecular motion is isotropic;
3) molecular motion and the dynamics of the electron magnetic moment are uncorrelated;
4) the nuclear relaxation correlation time is much shorter than the nuclear relaxation time, the electron relaxation correlation time is much shorter than the electron relaxation time (Redfield limit);[41]
5) the electronic g-tensor is isotropic, and the static Hamiltonian is described exclusively by the Zeeman interaction without taking into account anisotropic effects.
Approximations 1 – 3 appear to be quite reasonable in the sense that deviations from them do not cause significant changes in the relaxation enhancement at room temperature.[25][44][45] The anisotropy of the g-tensor of the nucleus and electron also does not lead to significant effects,[46-48] and deviations from the Redfield limit can significantly affect the relaxation times.[49] Under the conditions of approximation (see Eq. (5)), anisotropic effects, such as the ZFS or the anisotropy of the HFC tensor, can have a significant impact on relaxation times. To account for them, appropriate terms must be introduced into the static Hamiltonian. Anisotropic effects, one way or another, lead to an angular dependence of the relaxation enhancement effect, and this effect is described by including spherical harmonics in the expression.[50] Anisotropic effects are extremely important to consider for metal ions exhibiting high magnetic anisotropy, as well as for all single-molecule magnets.
In general, the theory of nuclear relaxation in paramagnetic molecules is quite complex. This review provides a general outline of the relevant effects. A more in-depth study of the nuclear relaxation effect is available in the work[41].
3. Practical applications of NMR spectroscopy in studies of the structure and properties of paramagnetic compounds
3.1. NMR spectroscopy of paramagnetic compounds and the method’s scope
The strong enhancement of relaxation for some metal ions hinders the observation of signals in NMR spectra. Due to the weak magnetic anisotropy of such ions, signals from different nuclei cannot be distinguished, therefore, the applicability of this method to them is limited. Fig. 3 compares the effects of signal broadening and dipole hyperfine shift. As can be seen from the figure, significant dipole shifts can be observed outside the ‘blind’ zone for some metal ions, e.g., for the lanthanides, with the exception of EuII and GdIII, ions, as well as for NiII, FeIII, FeII, CoII-HS, where HS denotes the high-spin state. For other ions, using dipole shifts to study structure and properties can be problematic without optimizing experimental conditions, which may include changing temperature and the external magnetic field.
![[{"id":"XsrGDrXlSZ","type":"paragraph","data":{"text":"Comparison of the effects of signal broadening and dipole hyperfine shift for metal ions. The red circle indicates the ‘blind’ region within which the <sup>1</sup>Н signal broadening reaches 100 Hz; the blue line indicates the region within which the dipole shift reaches 1 ppm. The calculations were performed by the authors of the review for the axially symmetric case by Eqs (38), (47), (51) using the characteristic values of Δχ<sub>ax</sub> and τ<sub>e2</sub> at a temperature of 298 K and a magnetic field of 18.8 T (NMR spectrometer operating at a proton frequency of 800 MHz)."}}]](/storage/images/resized/CuuWfxt1Cf9gc1qjt6VaRYSOdiBOZYKZwPhdiPIg_xl.webp)
Another large class of paramagnetic compounds are free radicals. These species are widely studied by EPR spectroscopy due to their relatively slow relaxation time (~10–7 s).[51] According to theoretical calculations,[52] the expected signal width for nitroxyl radicals in NMR spectra is approximately 100 – 1000 Hz, which corresponds to the threshold values for its detection. Consequently, they are poorly suited for NMR spectroscopy. Nevertheless, the authors of some studies[34][52-54] managed to record and analyze NMR spectra for a number of nitroxyl radicals. In particular, simulated and experimental spectra for 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (4-hydroxy-TEMPO) are shown in Fig. 4.
![[{"id":"CoYgtfkuAs","type":"paragraph","data":{"text":"Simulated (<i>a</i>) and experimental (<i>b</i>) <sup>1</sup>Н NMR spectra of 4-hydroxy-TEMPO radical (600 MHz) at 290 K.<sup>52</sup> Copyright © Wiley-VCH Verlag."}}]](/storage/images/resized/yHtFxuxY8Y3nqBmyMuTpPJXCHIX6qEeU4Cjxuebv_xl.webp)
In general, the number of studies devoted to the NMR of free radicals is extremely limited due to the complexity of data acquisition and interpretation, as well as their low information content. However, the combination of a paramagnetic metal and a ligand containing an unpaired electron (radical) can result in the recording of an NMR spectrum with higher resolution than that of a diamagnetic metal. When an organic radical combines with a paramagnetic metal, the unpaired electrons on the metal influence the relaxation of the unpaired electron on the ligand. This increases the rate of electron relaxation and, in turn, reduces the line broadening of the NMR nuclei.
Consequently, the NMR detectability of organic radicals can be improved by their coordination with paramagnetic metals. For example, complexes of anionic Ph-BIAN ligands (BIAN is bis(phenylimino)-acenaphthenequinone) with paramagnetic lanthanide ions were obtained (Fig. 5).[55] Similar conclusions that the NMR detectability of organic radicals can be improved by their coordination with paramagnetic metals were made for a series of phthalocyanine complexes of yttrium, terbium, and dysprosium(III).[56]
![[{"id":"wDL5Otcekz","type":"paragraph","data":{"text":"<sup>1</sup>Н NMR spectra of solutions of complexes Ln(Ph-BIAN)<sub>3</sub> (Ln = Tb<sup>3+</sup>, Dy<sup>3+</sup>, Ho<sup>3+</sup>, Er<sup>3+</sup>, Tm<sup>3+</sup>) in toluene-d<sub>8</sub>, measured in a magnetic field of 15.1 Т at 296 K. The insets demonstrate RQC splitting of the <sup>2</sup>H NMR signal in partially deuterated analogs. The colored lines illustrate the shifts in signals from identical nuclei."}}]](/storage/images/resized/OZhLdxJfTsmW7JFW9qCROL3OawkR8hGQXETkTUDw_xl.webp)
Chemical exchange processes (ligand exchange, tautomerism, solvent coordination, etc.) in paramagnetic systems present a particular challenge. They lead to the emergence of several chemical or coordination forms of the molecule, in which the nuclei under study are located in different local magnetic fields generated by the unpaired electron. This leads to averaging of signals by the exchange mechanism at intermediate or high rates relative to the NMR time scale, resulting in extreme line broadening, even to the point of their complete disappearance. Thus, for systems with fast chemical exchange involving nuclei near the paramagnetic center, obtaining a resolved NMR spectrum is often impossible without altering the experimental conditions (e.g., lowering the temperature to slow down the exchange).
However, even for paramagnetic compounds, in the absence of rapid chemical exchange, recording spectra is not a trivial task. Enhanced relaxation results in low signal intensity, especially for heteronuclear nuclei (e.g., 13C) under conditions of normal proton decoupling or for nuclei with low natural abundance. Therefore, to record spectra of paramagnetic compounds, it is necessary to use the highest possible concentrations (usually in the range of (2 – 10) × 10–2 mM and higher) to compensate for the decrease in signal intensity.
The acquisition time (AQ) and relaxation delay (d1) in the NMR pulse sequence should be selected according to the relaxation characteristics of the sample. The optimal approach is to pre-estimate the longitudinal (T1) and transverse (T2) relaxation times for key signals. It can then be established that AQ ≈ 5T2, d1 ≈ 5T1. For typical paramagnetic complexes, T1 and T2 values range from fractions to tens of milliseconds. Accordingly, the optimal AQ and d1 are often at the minimum limit technically acceptable for modern NMR spectrometers (usually on the order of 10 – 100 ms).
The choice of magnetic field, duration, and power of the excitation pulse also require attention. Using a strong magnetic field and a 90° pulse theoretically ensures maximum coherence and, therefore, signal intensity. However, the spectra of paramagnetic compounds are characterized by an extremely wide range of chemical shifts (hundreds, thousands, and even tens of thousands of ppm). When using a short, broadband pulse, excitation becomes highly non-uniform: signals far from the center of the spectrum are excited significantly less strongly. This leads to two key problems:
1) the impossibility of correctly quantitatively integrating signals located in different parts of the spectrum;
2) a decrease in intensity, up to complete disappearance, of signals at the edges of the spectral window.
To ensure more uniform excitation, it is necessary to shorten the pulse duration and, consequently, increase its excitation bandwidth. However, the same pulse in a weaker magnetic field will cover a wider ppm range, making the excitation more uniform. Therefore, the optimal magnetic field and pulse duration must be selected individually for each sample based on the expected shift range. For the convenience of researchers, the authors of this review developed a special online tool that allows for calculating the optimal pulse duration based on the expected spectral window.[57]
Regarding two-dimensional NMR techniques, many of them prove inapplicable to paramagnetic systems due to the rapid loss of spin coherence during the pulse sequence. Experiments with minimal evolution times, such as two-dimensional (2D) NMR techniques such as Correlation Spectroscopy (COSY) and Heteronuclear Single Quantum Coherence (HSQC), prove to be the most viable. A detailed analysis of the applicability of 2D techniques to paramagnetic complexes is presented in the study.[58]
Signals from different groups of nuclei often overlap in the spectra of paramagnetic complexes. An effective method for resolving these overlaps is to slightly change the spectrum recording temperature. Since contact and pseudocontact shifts often have different temperature dependences, the total shift for nuclei with different ratios of these contributions will change differently. As a result, changes in temperature can separate the overlapping signals, allowing each to be identified and analyzed.
After successfully recording a spectrum, the researcher is faced with the task of separating the total paramagnetic shift into diamagnetic, contact, and pseudocontact components. Direct use of the contact shift to obtain structural information is limited. The most striking example is the fact that the contact shift is a direct experimental indicator of the degree of covalence of the chemical bond between the paramagnetic center and the ligand.[59] In turn, the pseudocontact shift, determined by the geometry of the complex and the anisotropy of the magnetic susceptibility, is the main source of information for structural studies in solution and the determination of the parameters of magnetic interactions.
3.2. NMR spectroscopy as a method for studying precursors of magnetic materials
3.2.1. Structural studies of metal complexes
Studies of molecular structure by pNMR technique are based on the dependence of certain paramagnetic effects on nuclear coordinates. Primarily, this is the dipole hyperfine shift, which depends on spherical or polar (in the case of axial symmetry) coordinates. To use the dipole shift for this purpose, its contribution must be separated from other effects viz., contact and diamagnetic. Another effect is relaxation enhancement, the magnitude of which is influenced only by the distance between the nucleus and the electron localization center, within the relevant assumptions (see Section 2). The RDC and RQC effects also depend on the spherical or polar coordinates of the bond vectors between a pair of specific nuclei.
Objects for structural studies using pNMR can be divided into two groups: macromolecules and small molecules. In the former case, the concept of paramagnetic labels is used, introduced into the molecule under study, and the induced paramagnetic effects upon label introduction help to establish the spatial structure of the macromolecule. Paramagnetic labels are typically used to establish the structure of proteins.*
As for the objects of the second group (small molecules), structural studies of paramagnetic metal complexes are based on the structure of the ligands coordinated to the paramagnetic ion or their intermolecular interactions with the paramagnetic molecule. One of the simplest and most effective ways to detect complex formation between a metal ion and a ligand is by analyzing NMR spectra. This approach is more often used for diamagnetic complexes, but is more effective for paramagnetic ones. This is because a paramagnetic metal ion has far more impact on the characteristics of the ligand signal than a diamagnetic one. Moreover, the quantitative change in these characteristics depends on the spatial arrangement of the ligand nuclei, paving the way for elucidation of the structure.
Often, the goal is not to precisely determine the structure of a complex in solution, but rather to merely capture complex formation and, possibly, identify binding sites. Similar problems occur frequently in studying the mechanisms of chemical reactions that include paramagnetic metal complexes as catalysts.[60] Much of this work involves investigating the effect of paramagnetic enhancement of substrate nuclear relaxation due to their intermolecular interaction with a paramagnetic ion. This interaction results in broadening of the signals from the substrate nuclei, which is quantitatively analyzed within the framework of the Solomon – Bloembergen – Morgan theory to estimate interatomic distances in solution.[61-63]
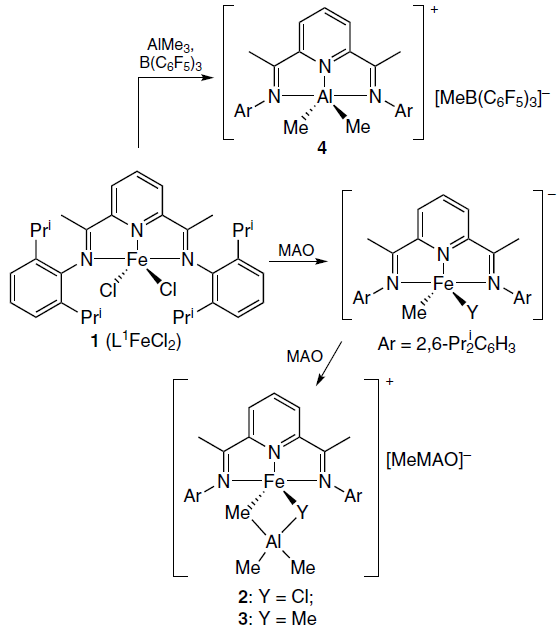
A number of works are devoted to NMR studies of the mechanisms of activation of metallocene iron(II) and cobalt(II) catalysts in olefin polymerization reactions under the influence of various trialkylaluminum compounds.[64-66] Fig. 6 shows the changes in the 1H NMR spectrum of one of the metallocene catalysts 1 (L1FeRR') in the presence of various activators, viz., methylalumoxane (MAO) with different ratios of Al and Fe ions, as well as the AlMe3 – B(C6F5)3 system.[67] Thus, the structure of intermediate and final complexes 2 – 4, the products of the reaction of catalyst 1 with MAO and AlMe3 – B(C6F5)3 (Scheme 1), was determined. Similar studies were carried out for cobaltocene[68][69] and vanadocene catalysts.[70]
![[{"id":"svHjPjtYy2","type":"paragraph","data":{"text":"NMR spectra (toluene-d<sub>8</sub>, 296 K) of L<sup>1</sup>FeCl<sub>2</sub> (1) prior to (<i>a</i>) and after reaction with different activators: methylalumoxane at a ratio of Al : Fe = 20 (<i>b</i>) и 50 (<i>c</i>); as well as with AlMe<sub>3</sub> – B(C<sub>6</sub>F<sub>5</sub>)<sub>3</sub> at a ratio of Fe : Al : B = 1 : 15 : 1.1 (<i>d</i>). <sup>67</sup> Copyright Taylor and Francis Group, 2017."}}]](/storage/images/resized/WPEo4l0VBrDlYF9sulbjOdEg2oPntH5prMlxQKL0_xl.webp)
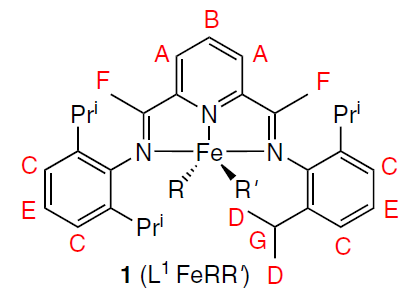
Modeling the various components of the chemical shift of nuclei in 3d-element complexes is approached in different ways. The diamagnetic component is usually taken from NMR spectra of a structurally similar diamagnetic metal complex[66] or a free ligand.[71] Typically, the diamagnetic component is small compared to the paramagnetic component, so any errors that arise do not significantly affect the results. Some authors (see, e.g., Ref. [72]), however, note the influence of this error in individual cases. The contact shift is calculated using quantum chemistry methods, and it is here that the majority of the error in this method is concentrated. Much effort is being expended to improve the accuracy of contact shift calculations,[65][66][73] and currently developed methods require significant computational resources. Dipole shifts are explicitly correlated with nuclear coordinates, so modeling this component of the chemical shift in the case of orthorhombic symmetry requires finding one or two anisotropy values of the magnetic susceptibility tensor. In the case of two or more magnetoactive nuclei in a molecule, determining the optimal anisotropy values is not difficult. In this way, the structures in solution of many transition metal and lanthanide complexes[74-77] of practical value have been confirmed: catalysts (including chromium(III),[11][78][79] cobalt(II),[71][80][81] and iron(II) complexes[71][81]) as well as single-molecule magnets (SMMs).[27][82][83]
For 4f elements, the relationship of the lanthanide-induced shift (LIS) with the structure of complexes has long been considered through the prism of Bleaney’s theory.[84] According to this theory, the dipole component of the LIS can be represented as a function of the crystal field parameter B02 for axially symmetric complexes
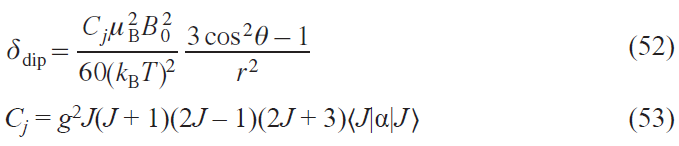
where Cj is the tabular constant for lanthanide ions in Bleaney’s theory.
The contact component was considered as a function of the hyperfine interaction constant Fk for the k-th nucleus, arising due to the delocalization of the spin density on the ligand, and the tabular value ⟨SZ⟩ for each of the lanthanide ions, which represents the average electron spin projection SZ of the Ln ion along the quantization axis
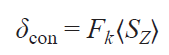
The Bleaney’s theory is based on a number of assumptions. For example, it is assumed that the splitting in the ligand field is significantly smaller than kBT and amounts to 205 cm–1 at 298 K. Furthermore, it is believed that higher-order crystal field parameters do not contribute to the LIS. Finally, it is assumed that in a series of isostructural complexes with various lanthanides, the magnetic axes of the lanthanide ions do not change their orientation.
Approaches to the analysis of NMR spectra were based on the study of series of predominantly isostructural complexes and the use of various linear forms that were constructed from combinations of Eqs (52) and (54) written for one, two or three resonating nuclei.[84] Perhaps the most successful was the use of the dual-nuclei method,[85] which made it possible to get rid of the crystal field parameter: thus, for a pair of resonating nuclei k and l, the following relationship was satisfied
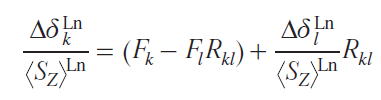
where Rkl is the ratio of geometric factors for k and l nuclei.
This linear form corresponds to a straight line in coordinates
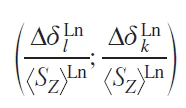
or a series of isostructural complexes with a slope equal to the ratio of the geometric parameters Rkl of these protons and a free term (Fk – FlRkl) close to zero due to the negligible values of the contact contributions to the LIS. The resulting Rkl characteristics can be used to determine the structure of the complexes in solution.
A significant drawback of this method is the need to synthesize a series of complexes with different lanthanides. Moreover, over time, it was shown that the assumptions of the Bleaney’s theory are not generally fulfilled,[86] leading to difficult-to-interpret kinks in dependences similar to Eq. (55).[87][88] As a result, in recent years, these approaches have been almost never used for the analysis of complex structures by NMR spectroscopy.
Currently, modeling chemical shifts in NMR spectra is used to solve such problems. The availability of information on diamagnetic shifts (ddia), structural information from X-ray diffraction data or quantum chemical modeling, and the ability to assign at least one signal in the spectrum to a paramagnetic complex lead to a simple relationship that allows the calculation of the positions of the remaining signals.
Thus, neglecting the contact components, the ratios of lanthanide-induced shifts for different pairs of protons Hk and Hl can be described with sufficient accuracy by the ratio of the corresponding geometric factors Rkl from Eq. (55), and Eq. (56) can be derived for estimating the positions of resonance signals in the spectrum of the paramagnetic complex
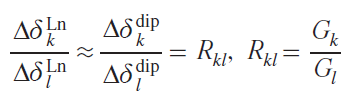
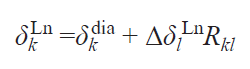
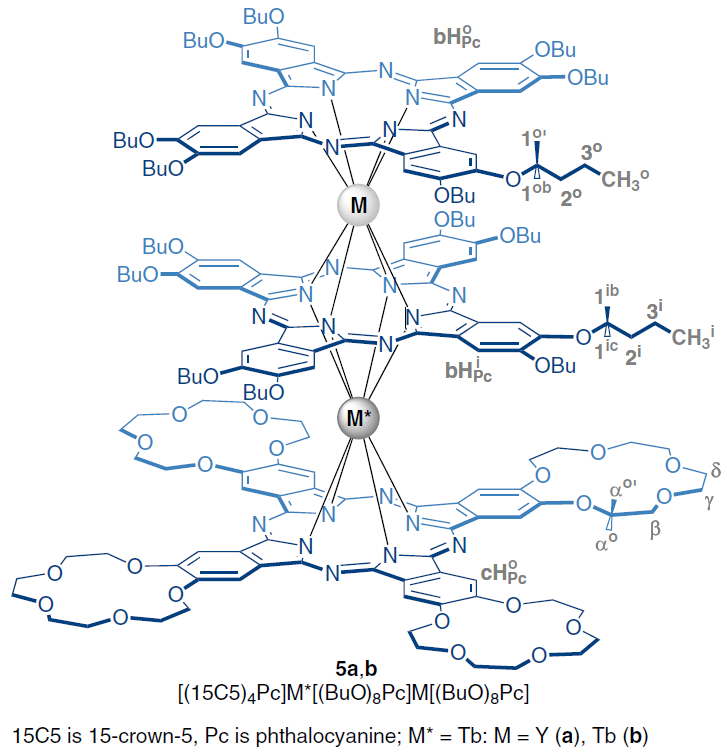
By substituting into Eq. (57) the value of the reliably established LIS for the Hl proton, as well as the magnitudes of the Hk proton resonance signals in the spectrum of the isostructural diamagnetic complex, and the corresponding ratios of the geometric parameters for Hk and Hl, one can find the desired position of the Hk proton resonance signal in the spectrum of the complex containing a paramagnetic lanthanide. This equation has been successfully used to analyze the spectra of paramagnetic sandwich complexes of lanthanides with phthalocyanine ligands, for example, heteronuclear yttrium and terbium trisphthalocyaninates 5.[89][90]
When signals shifted in different directions relative to the signals of diamagnetic reference compounds appear in the NMR spectra of such complexes, it is useful to construct contour maps of the function (3cos2q – 1)r–3 and plot the coordinates of nuclei of different structural types on them, which helps to interpret the observed spectral features (Fig. 7a). Good agreement between the experimental and simulated chemical shifts suggests that the structures of the complexes in solution and in solid form are similar. The reliability of the result is measured by the data discrepancy criterion R2: the closer it is to unity, the higher the reliability (see Fig. 7b). If R2 is far from unity, then the structures of complex 5a in solution and in crystal differ.
![[{"id":"TJCOZdzzkV","type":"paragraph","data":{"text":"Contour map of the geometric parameter function G(q,r) showing the position of different protons in regions with positive and negative LISs (<i>b</i>), as well as the <sup>1</sup>H NMR spectrum of complex <b>5a</b> in toluene and a comparison of the simulated and experimental values of the proton chemical shifts (<i>c</i>)."}}]](/storage/images/resized/eIH820vv4AtHin6Fp7rR2LqX6mhE7FCyOog1OBB0_xl.webp)
Two approaches have been developed for analyzing the spectra of heteronuclear lanthanide complexes containing nonequivalent paramagnetic metal centers. One is based on the synthesis and characterization of isomeric complexes in which one paramagnetic center is retained, while the others are replaced by diamagnetic rare-earth ions (La, Lu, Y). This method was used to analyze the spectra of heteroleptic trisphthalocyaninates of general formula [(BuO)8Pc]M*(Pc)M(Pc). It was found that the spectrum of homonuclear complexes M = M* = Ln (Tb, Dy, Ho, Er, Tm, Yb) can be represented as a superposition of the spectra of isomeric heteronuclear complexes with M = Y, M* = Ln and M = Ln, M* = Y.[38] Analysis of a series of spectra of heteronuclear complexes containing dia- and paramagnetic centers showed that the axial anisotropy of Ln3+ ions in a symmetric phthalocyanine environment (position M) was systematically higher compared to that for isomeric complexes with lanthanides in position M*.[91]
An alternative method that involves the preparation of isomeric heteronuclear complexes, is based on the additivity of lanthanide-induced shifts from different paramagnetic metal centers. As a result, the LIS can be represented as the sum of two contributions, determined by the geometric parameters of the resonating nucleus under consideration relative to each of the lanthanides and the corresponding axial anisotropies (χax)
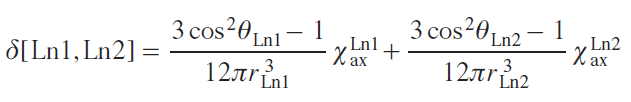
The search for a combination of χaxLn1 and χaxLn2 that provides the smallest deviation between the simulated and experimentally recorded NMR spectra allows not only to assign signals but also to simultaneously study the influence of the coordination environment on the magnetic properties of various metal centers. This approach was first used for the heteroleptic trisphthalocyanine [(15C5)4Pc]Tb[(BuO)8Pc]Tb[(BuO)8Pc], complex 5b, which undergoes a change in conformational state when changing the solvent.[92] Specific solvation of crown ether substituents by dichloromethane molecules results in the ligands in the (15C5)4Pc]Tb[(BuO)8Pc] fragment being in a staggered conformation with a square-antiprismatic environment of the Tb3+ ions, while the [(BuO)8Pc]Tb[(BuO)8Pc] moiety has a skewed conformation with a twist angle of about 22°. As a result, the anisotropies of the terbium ions in these fragments differ significantly, amounting to 8.70 × 10–31 and 9.18 × 10–31 m3, respectively. When switching from dichloromethane to toluene, the solvation type changes. This leads to neighboring phthalocyanine ligands adopting staggered conformations in pairs, and the anisotropies of the Tb3+ ions become virtually equal: 7.65 × 10–31 и 8.05 × 10–31 m3, respectively. For comparison, isomeric heteronuclear complexes containing a pair of Tb3+ and Y3+ ions were synthesized; analysis of their spectra confirmed the observed spectral correlations.[90] Similar conclusions were made for heteronuclear complexes with Tb3+ and Dy3+ ions.[93]
An analysis performed for homoleptic triple-decker crown phthalocyaninates of rare earth elements showed a twofold increase in axial anisotropy upon moving from dysprosium(III) complexes to terbium(III) complexes. A study of the temperature dependences of the chemical shifts of proton resonance signals in the spectra of these complexes showed that terbium complexes exhibit the highest temperature sensitivity (∂δ/∂T = 1.1 ppm/K).[94] This fact demonstrates the relationship between the fundamental magnetic properties of the complexes and the potential for their practical application as NMR thermosensitive contrast reagents.[95]
* There are several approaches to introducing a paramagnetic label into a macromolecule, but this area is beyond the scope of this review.
3.2.2. Single-molecule magnets
Molecular magnets[96][97] are bistable compounds that exhibit the properties of macroscopic magnets even at the level of a single molecule. The main property of SMMs is the retention of magnetization acquired in a magnetic field after the field is switched off, i.e., a single molecule behaves like a permanent magnet. These unique properties of SMMs determine the prospects for their practical application in the creation of high-density information storage devices at the molecular level,[98] molecular spintronics[99] and magneto-optics,[100] as well as qubits in quantum computers.[101]
When a SMM is exposed to a magnetic field, the spin magnetic quantum number MS takes on the most energetically favorable value –S, which corresponds to the orientation of the magnetization vector ‘along the field.’ After the field is switched off, the magnetization thus induced can persist for some time due to the presence of an energy barrier U for magnetization reversal for the process of changing the orientation of the magnetization vector.
The main necessary condition for the existence of the barrier U is a negative value of the splitting energy in zero field D (according to the SH formalism), since it is in this case that the direct transition between the states MS = –S and MS = +S (or MJ = –J and MJ = +J ) is forbidden by the selection rules for the magnetic dipole moment in the first order, and for the relaxation of magnetization (the transition MS = –S → MS = +S ) it is necessary to overcome the U barrier. The highest magnetization reversal barriers are demonstrated by lanthanide complexes, therefore it is this class of compounds that is most interesting for use as SMMs.
The ground-state electronic structure of a free lanthanide ion is a set of degenerate (2J + 1) levels (where J is the quantum number of the total angular momentum of the electron). The crystal field effects of ligands that arise in metal coordination complexes are generally significantly weaker than the spin-orbit interaction, since the atomic f-orbitals of the lanthanide ion are only slightly involved in the formation of bonding molecular orbitals. Nevertheless, it is the crystal field that removes the degeneracy of the (2J + 1) levels and thus determines the magnetic properties of the compound. Currently, the various magnetic properties of lanthanide complexes are widely in demand for a variety of practical applications, including SMMs,[96] paramagnetic probes,[102][103] shift and relaxation agents,[104][105] in vivo temperature and pH sensors in MRI,[106][107] elements of quantum computers.[99] Therefore, studying the field parameters of ligands of lanthanide complexes is an important task from the point of view of the above-mentioned areas of their practical application.[108][109]
A traditional method for determining the structure of the electron levels of lanthanide complexes is luminescence spectroscopy.[110][111] The fine structure of bands in luminescence spectra arises due to splitting of the ligands by the crystal field and, accordingly, can be used to determine the electron levels of the ground state. However, in many cases the signal in the luminescence spectrum is not sufficiently resolved to reveal the fine structure, and in the case of complexes with ligands possessing high extinction coefficients, f-luminescence may not be observed in principle, which makes this approach inapplicable for studying SMMs.
Determination of the electron levels of lanthanide complexes using NMR spectroscopy becomes possible due to the preparation of isostructural complexes with diamagnetic metal centers (lanthanum, yttrium or lutetium) and neglect of the contact component of the lanthanide-induced shift (with the rare exception[112]). Taking these factors into account, it is possible to find with high accuracy the values of the dipole components of the shifts, directly proportional to Δχax, indirectly related to the value of the energy barrier U. In turn, under the condition of thermodynamic and kinetic stability of the complexes in solution, the proportionality coefficients between Δδdip and Δχax, which are geometric parameters of G = (3cos2q – 1)r–3, are taken from the X-ray structural data or the results of quantum-chemical modeling (see Eq. (57)). Therefore, this type of analysis is most often used for complexes with rigid macroheterocyclic ligands, such as tetrapyrroles (see Refs [89][90][93][113] as a continuation of the work of Ishikawa and co-workers[12]). Examples of the application of similar studies to endohedral complexes of lanthanides with fullerenes are also described.[114][115]
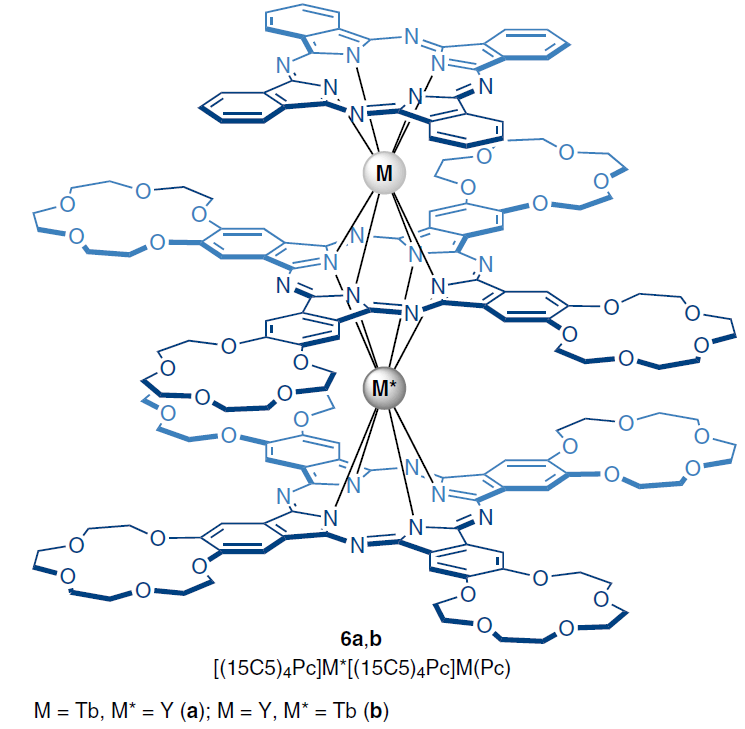
Further linearization of the LIS dependences on geometric parameters allows us to find the values of the axial component of the magnetic susceptibility tensor Δχax and investigate the relationship between the structure of the complexes and their magnetic properties. Among the paramagnetic lanthanide complexes studied by NMR spectroscopy and exhibiting slow magnetization relaxation, heteroleptic heteronuclear trisphthalocyaninates 6a,b of the general formula [(15C5)4Pc]M*[(15C5)4Pc]M(Pc), where M, M* = Tb, Y, should be mentioned.
Thus, a comparison of the values of the U barrier and the parameter Δχax for two heteronuclear complexes 6a,b with ions M*, M = Tb, Y showed that a decrease in the symmetry of the coordination environment of the Tb3+ ion in the M position leads to an increase in both the anisotropy (8.94 × 10–31 m3), and the magnetization relaxation barrier (168.1(8) K at 1500 Oe) compared to a more symmetric environment of the Tb3+ ions in the M* position (Δχax = 8.41 × 10–31 m3; U = 129.8(0) K at 1500 Oe).[89][116]
A unique air-stable heteroligand sandwich terbium p-complex (TPP)Tb(Cp*) (7), where TPP is tetraphenylporphyrinate dianion, Cp* is pentamethylpentadienyl, was studied using magnetometry and NMR spectroscopy techniques.[112]
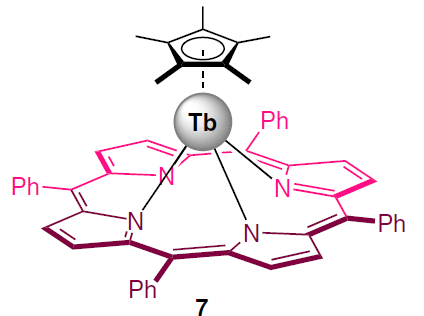
Quantum-chemical calculations revealed a significant contact contribution to the LIS values of the protons of pentamethylpentadienyl ligands, separated from the Tb3+ ion by only three chemical bonds. Subtracting the calculated contact contributions from the LIS values allowed us to find the dipole components, from which the axial anisotropies were then determined. It was found that complex 7 is characterized by significantly lower values of both the anisotropy Δχax (6.30 × 10–31 m3), and the barrier U (58.3 K at 1000 Oe) compared to classical sandwich terbium complexes with tetrapyrrole ligands. The low value of the axial anisotropy is consistent with the results of quantum chemical calculations performed using the complete active space self-consistent field (CASSCF) method. Calculation results showed that such a ligand environment does not cause significant splitting of the energy levels responsible for the slow relaxation of magnetization.
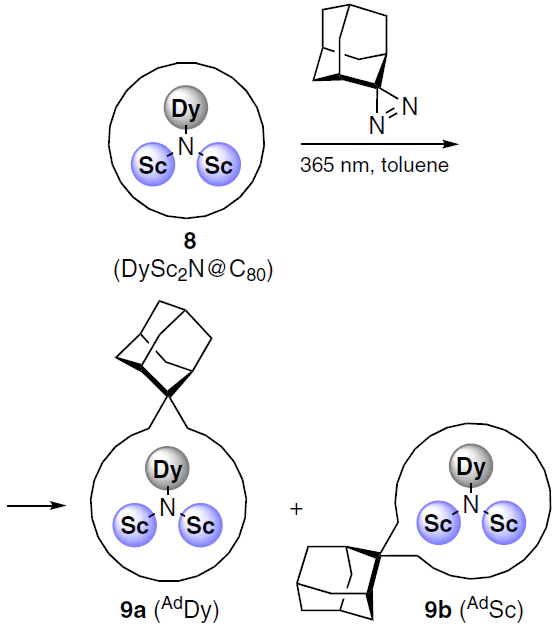
Interesting relationships between structural, spectral and magnetic characteristics were found for the DySc2N cluster encapsulated in C80 fullerene using quantum chemistry methods.[114]
The authors carried out photochemical addition of adamantylidene, a carbene derived from 2-adamantane-2,3'-[3H]diazirine, to the endohedral fullerene complex DySc2N@C80 (8) (Scheme 2). The reaction afforded products of the carbene insertion in the bonds between two six-membered ([6,6]) or five- and six-membered ([5,6]) rings. The cluster could orient itself within the fullerene cavity such that either the dysprosium or scandium atom was located near the adamantyl fragment, corresponding to the AdDy (9a) or AdSc (9b) structures. The structure of products 9a,b could be characterized due to the paramagnetism of dysprosium in the endohedral cluster, and it was found that the product [6,6]-AdSc is characterized by both higher anisotropy (1.78 × 10–31 m3) and a higher blocking temperature (7.9 K) compared to the isomer [6,6]-AdDy, the analogous parameters of which were 1.60 × 10–31 m3 and 4.6 K. The blocking temperature of the starting metallofullerene DySc2N@C80 was 6.9 K.
An alternative method for determining Δχax is based on the analysis of residual quadrupole couplings in anionic sandwich complexes of lanthanides Ln(tdnCOT)2– (10) with deuterated cyclooctatetraenide ligands. It also allowed the calculation of crystal field parameters using the temperature dependences of Δχax.[12]
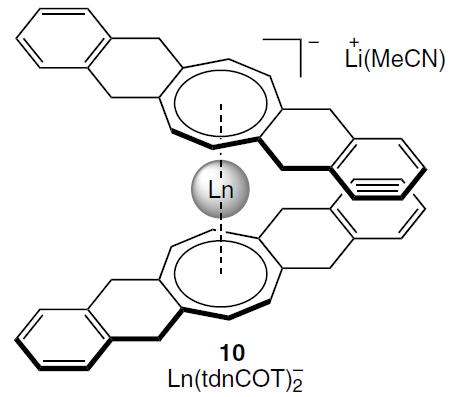
These approaches to structural analysis of lanthanide complexes can be extended to other nuclei. For example, in addition to 1H and 2H NMR spectroscopy for paramagnetic complexes, 31P NMR spectroscopy has been successfully used in the case of distorted trigonal bipyramidal complexes with tri(silyl)phosphide ligands [Ln{P(SiMe3)2}3(THF)2].[117]
Unlike lanthanide complexes, 3d transition metal complexes are less suitable for NMR studies for a number of reasons. Firstly, the nuclear relaxation times for many transition ions are much shorter, making the spectra less resolved. Secondly, the periodicity of properties in lanthanide complexes allows for a reduction in the number of unknown parameters in models describing experimental NMR spectra. Thirdly, since the electronic properties of transition metal ions vary greatly, complexes of different metals are rarely isostructural. Fourth, the significant contribution of d-orbitals of the metal ion to the molecular orbitals (MOs) of the complex means significant delocalization of the spin density, leading to significant (sometimes dominant) contact shifts, the error in calculating which using quantum chemistry methods can be quite large.[13][118] All this explain the fact that works devoted to the study of paramagnetic complexes of transition metals by NMR spectroscopy are quite scarce.[11][66]
In the case of low anisotropic ions (CrIII, MnII, CuII, VIV, etc.), for which contact shifts predominate, the problem of the inaccuracy of their quantum-chemical calculation comes to the fore.[119] For such ions, at the moment, no method has been found by which one can accurately determine the parameters of the electronic structure using NMR spectroscopy data.[11]
In general, the approaches to modeling experimental chemical shifts in NMR spectra for complexes of 3d-metal and lanthanide ions are similar, with the only difference being that the forms of the used SHs may differ (see Section 2.3).
An illustrative example of the successful application of NMR spectroscopy is the study of an iron(II) complex,[120] which exhibits a record-breaking proton chemical shift (~ 10 000 ppm). Using terahertz EPR spectroscopy, magnetometry, and NMR spectroscopy, the authors were able to determine the electron relaxation rate. Moreover, they showed that due to the large rhombicity of the system, the relaxation of nuclei associated directly with the iron(II) ion is suppressed, which makes it possible to register their signals even with such large shifts.
In some of the above-described examples of SMM studies based on both 3d and 4f metal ions, it can be noted that NMR spectroscopy is used in a magnetic field, along with other methods. This trend is not accidental, as the target characteristics of electronic structure and magnetic interactions can rarely be measured directly. As shown above, they are determined indirectly, by describing experimental data from methods using specific models in which the desired quantities serve as parameters. This approach always relies on the adequacy of the model and the unambiguity of the modeling results. However, there is almost no examples of assessing the reliability of modeling data from magnetic methods for determining specific physical quantities.
It was shown that when analyzing a limited set of experimental data within the framework of a SH model with a large number of parameters, the problem of overparameterization arises.[15] The existence of this problem has led to an increase in the number of studies in which the parameters of magnetic interactions are established by modeling experimental data obtained by several methods.[11-16] Of course, this is valid if it is proven that the structure of the compound being studied is the same for all samples. However, metal complexes often have different structures in the crystalline state and in solution, in which case this approach is invalid.
NMR spectroscopy and magnetometry methods were first used in 2003 by Ishikawa et al.[39] to determine the ligand field parameters for phthalocyanine anionic complexes of lanthanides — the first representatives of SMMs based on lanthanide complexes.[121][122] Since the Orbach magnetization reversal barrier directly depends on the energy levels, which are determined by the energy operator, the authors used the SH, which describes the ligand field parameters through Stevens operators (see Eq. (27)).[123] This approach has not undergone significant changes to date. Experimental NMR chemical shifts are related to the ligand field parameters in the SH by calculating the dipole component of the paramagnetic shift and the anisotropy values of the magnetic susceptibility tensor (see Section 2.3).
For different types of ligand crystal field symmetry, the set of parameters will differ. For example, in the case of complexes with D4d symmetry, the SH is significantly simplified
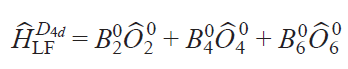
where Ô20 are Stevens operators.
In addition, the authors made an important assumption that all field parameters are linearly dependent on the number (n) of f-electrons in a series of isostructural lanthanide complexes, from f 8(Tb3+) to f 13(Yb3+)
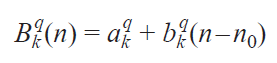
where n0 is the average number of f-electrons in the series of ions under study, Bkq(n) are the coefficients in front of the Stevens operators, k = 0, 2, 4, 6; q = –k, –k+2, ... , k.
The authors of the cited work do not strictly prove the validity of this assumption and are based on the fact that in the lanthanide series, the field parameters should vary monotonically depending on the number of f-electrons. This assumption, however, is important for the accuracy of determining the field parameters, since it reduces the number of unknown parameters from 3N (N is the number of isostructural complexes) to 6 and allows one to solve the problem of overparameterization shown in works[124][125]. It should be noted that this assumption is questioned by some authors.[126]
The essence of this approach lies in multidimensional optimization of crystal field parameters that best describe experimental data obtained by NMR spectroscopy and magnetometry. These two methods are related in solving this problem, as they allow for the experimental determination of certain components of the magnetic susceptibility tensor χ. While powder magnetometry for this tensor yields an isotropic value of χ– (see Eq. (61)), the NMR spectra contain information on its anisotropy — the quantities Δχax, Δχrh (see Eq. (38)). The crystal field parameters are optimized until the best fit between the experimental and calculated values (according to magnetometry data) and Δχ (NMR spectra) is achieved.
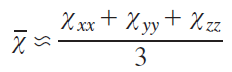
It should be noted that the authors neglected the influence of the contact shift for LnPc2– complexes.[39] Indeed, due to the weak participation of f-electrons in the formation of metal – ligand bonds (compared, e.g., to d-electrons of transition metal ions), significant delocalization of the spin density to the ligand nuclei does not occur.[9] However, it has been shown that for some lanthanide complexes (including phthalocyanine complexes)[87] the contact contribution to the chemical shift can be significant.[127]Thus, the issue of taking into account the contact contribution for lanthanide complexes is still a raging debate.
Importantly, Ishikawa et al.[121] found that using data from only one of the two methods also leads to the problem of overparameterization, i.e. the experimental data are equally well described by a fairly wide set of parameters, which makes it impossible to accurately determine the energies of the electron levels. In particular, this leads to errors in determining ligand field parameters ranging from 22% to 60% based on magnetometry data and from 5% to 20% based on NMR spectroscopy data. Only when modeling using both methods simultaneously does the parameter spread decrease sharply (the corresponding errors ranged from 1% to 3%), demonstrating the reliability of the results. The authors demonstrate the important role of NMR spectroscopy in studying the electronic structure of lanthanide complexes.
Modern studies utilize a combination of not only NMR spectroscopy and magnetometry, but also EPR spectroscopy in various frequency ranges, including the terahertz range (THz-EPR).[11][14] This hybrid approach allows for the reliable determination of magnetic interaction parameters. For example, Pavlov et al.[15] showed that only by combining magnetometry, X- and Q-band EPR spectroscopy, and THz-EPR can the SH model reliably describe experimental data.
3.2.3. Application of NMR in investigating spin transitions
Transition metal ions with d 4 – d 7 electron configurations can exist in states characterized by different values of the total electron spin moment (spin states). Different spin states of transition metal ions in complexes are characterized primarily by different electronic structures, i.e., different electron density distributions within the molecule. As a consequence, the spatial structure of such states also varies. Under certain external circumstances, a spin transition — a process that changes the spin state of a metal ion — may occur. Spin transition is considered a useful effect for practical applications (e.g., in the creation of sensors,[128][129] MRI agents,[130] ‘smart’ materials,[131] and memory devices),[132] and therefore attracts the attention of scientists in the fields of chemistry, physics, and engineering.
Several methods for studying the spin state of metal complexes are known based on the experimental measurement of a certain characteristic that varies for the proposed spin states. The most widely used method is magnetometry, which measures the magnetization of a sample, a characteristic directly related to the effective magnetic moment of the molecule. For many metal complexes, it is the spin angular momentum that makes the major contribution to the total orbital angular momentum, making it trivial to distinguish spin states using magnetometry. Other methods include X-ray diffraction analysis,[133] optical spectroscopy,[134] IR spectroscopy,[135] Mössbauer spectroscopy,[62] and EPR spectroscopy.[63]
NMR spectroscopy, in the form of the Evans method, is also used to study the spin state. Similar to magnetometry, it can measure the magnetization of a sample (see Section 2.1). Another approach, first used in 1965,[64] involves direct observation and analysis of the signals of the complex being studied in NMR spectra. In this work, a tetragonal nickel(II) complex was investigated and it was proposed to describe the contact shifts observed for it taking into account the population of both spin states of the nickel(II) ion: low-spin (S = 0) and high-spin (S = 1)
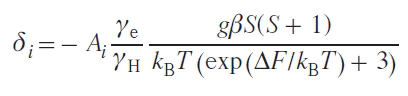
where Aiso is the isotropic HFC constant, ΔF is the change in free energy between states with S = 0 and S = 1. The authors assumed that the populations of spin states obey the Boltzmann distribution, and the process of changing the spin state at the molecular level is fast on the NMR time scale, which leads to a weighted average chemical shift
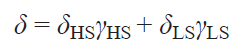
where γHS, γLS are fractions of high-spin (HS) and low-spin (LS) states.
Typically, the HFC value cannot be determined experimentally, therefore, despite their fundamental nature, Eqs (62) and (63) have little practical utility. The only way to theoretically estimate the HFC constant is through quantum chemical calculations.[65] This approach allows modeling the observed chemical shifts by the Eq. (62).[66][136]
Subsequently, a number of other approaches based on experimental data were proposed. One of these relies on empirical correlations between the spin state and observed chemical shifts in NMR spectra for a particular class of compounds. For example, a correlation was found between the chemical shift in the 15N NMR spectrum of a free ligand and the spin state of the corresponding iron(II) complex.[137] Moreover, the key chemical shift value can be calculated quite accurately using quantum chemical calculations using density functional theory (DFT). Therefore, this approach is viable even in the absence of experimental NMR spectra. Since chemical shift depends largely on the electron density distribution, the next logical step was to discover the dependence of the spin state on the effective charge of the nucleus forming the coordination bond with the metal ion.[138][139] A correlation was also found between the spin state and the chemical shift value of the nucleus of the complex per se.[140]
Obviously, the disadvantage of such approaches is their limited applicability, i.e., they can only be used for compounds of a certain class. A more general approach involves analyzing the temperature dependence of the observed chemical shift of the complex during the analysis of the temperature-induced spin transition. This approach has been actively used to study iron(II) complexes since the 1980s.[141] It is based on the hypothesis of a linear dependence of the paramagnetic shift on the inverse temperature, which follows from Curie’s law
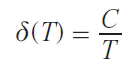
where C is the Curie constant.
In this case, a model of an ideal solution is used, in which the equilibrium constant between spin states and their populations obey the Boltzmann distribution[142]
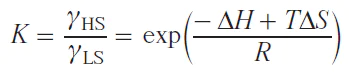
where ΔH, ΔS are enthalpy and entropy of the spin transition, respectively, R is the universal gas constant.
Considering that the low-spin state of the iron(II) ion is diamagnetic, the temperature dependence of the observed chemical shift is given by the Eq. (66):[143]
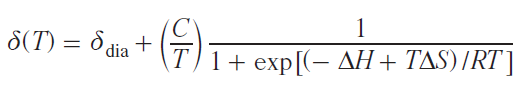
The dependence of proton shifts in the NMR spectrum of an iron(II) complex (see Eq. (66)) exhibiting a spin transition can be quite complex (Fig. 8).
![[{"id":"EAYax3kWyo","type":"paragraph","data":{"text":"Theoretical temperature dependences of the chemical shift in the NMR spectra of the iron(II) complex in the low-spin (blue curve) and high-spin (red curve) states. The case of a spin transition (green curve) was calculated by Eq. (66) using the following parameters: ΔH = 10 kJ mol<sup>–1</sup>, ΔS = 40 J (mol K)<sup>–1</sup>."}}]](/storage/images/resized/z70aEb6owPTvlqRBdFZbj9T5ZjFeci9XTmSFJnWr_xl.webp)
Using this approach, the spin equilibria of various iron(II) coordination compounds were studied.[141][143-145] However, for some complexes, this approach proved inapplicable, which created the prerequisites for its modification. The reason for the inapplicability of the above-described approach is its deviation from the linear Curie dependence (44), which is valid for an isolated electron. For the iron(II) ion, the high-spin state is characterized by four unpaired electrons, so the presence of anisotropic splitting effects in zero field and/or ‘admixture’ of low-lying excited states can lead to a deviation from the linear dependence.[146] Currently, these effects are taken into account in two ways. The first involves limiting the temperature range over which NMR experiments are conducted; this is typically no greater than 100 K. Within such a narrow range, deviations from linearity are often not observed. To account for deviations from linearity outside this range, a constant term is added, and the expression is transformed[147]
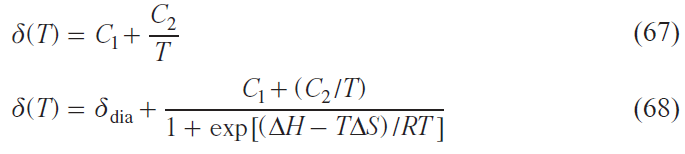
where the C1 and C2 coefficients, as in Eq. (69), represent the model parameters.
This approach has proved very effective for a wide range of iron(II) complexes.[147-149]
The second, more general method involves directly taking into account the excited state[146]
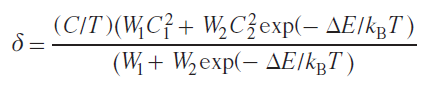
where W1 and W2 are the fractions of the ground and excited states; C1 и C2 are coefficients accounting for the spin density of the ground and excited states; ∆E is the energy difference between the ground and excited states. This method is described in more detail in the works[146][150][151] together with examples of its practical application.
The first method is essentially a shortened version of the second, in which only the first term of the expansion of the exponential function into a power series is taken into account.
For cobalt(II) complexes, a slightly different approach must be used. The low-spin state of the cobalt(II) ion (d7), unlike that of iron(II) (d6), is paramagnetic (S = 1/2), as is the high-spin state (S = 3/2). In this regard, the expression for the observed chemical shift of spin-transition cobalt(II) complexes will be as follows:
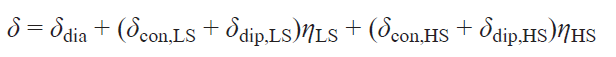
where η is the population of the corresponding spin state.
Considering that the anisotropy of the magnetic susceptibility tensor of the cobalt(II) ion in the low-spin state is much smaller than in the high-spin state, the dipole shift of the low-spin fraction can be neglected[106]
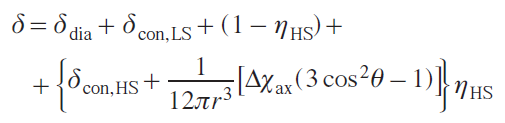
This is due to the total spin S = 1/2 for the LS state, which lacks anisotropic splitting effects at zero field, and to the absence of low-lying excited states. On the other hand, the HS state of the cobalt(II) ion typically exhibits the highest magnetic anisotropy among transition metals.[152] Furthermore, this leads to a deviation from Curie’s law, which is reflected in a deviation of the dipole shift dependence from linearity.[153]
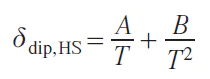
where A and B are parameters.
Therefore, for cobalt(II) complexes, it is proposed to describe the experimentally observed chemical shift value as follows:
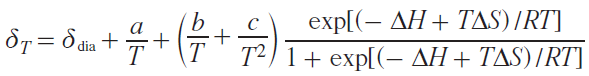
where a, b, c are model parameters.
A typical temperature dependence of the chemical shift for the spin-transition complex of cobalt(II) is shown in Fig. 9.
![[{"id":"Jdx1jH_Baa","type":"paragraph","data":{"text":"Theoretical temperature dependences of the chemical shift for the cobalt(II) complex in the low-spin (blue curve) and high-spin (red curve) states. The case of a spin transition (green curve) was calculated by Eq. (73) using the following parameters: ΔH = 12 kJ mol<sup>-1</sup>, ΔS = 40 J (mol K)<sup>–1</sup>."}}]](/storage/images/resized/8wYDILIlLfhWEcAqyjuatZcoJbRBnCTFHeTCoT9L_xl.webp)
A relatively small class of spin-transition complexes based on the iron(III) ion stands apart.[154] A distinctive feature of the iron(III) ion is that, as in the case of cobalt(II), both spin states are paramagnetic: both LS (S = 1/2) and HS (S = 5/2). However, the LS state of FeIII can exhibit significant magnetic anisotropy due to low-lying excited states, unlike CoII. Thus, the simplification made for cobalt(II) complexes regarding the insignificant dipole shift of the LS state is not applicable in the case of iron(III). Therefore, in the case of iron(III) complexes, the most general model should be used, taking into account all components of the chemical shift of both spin states
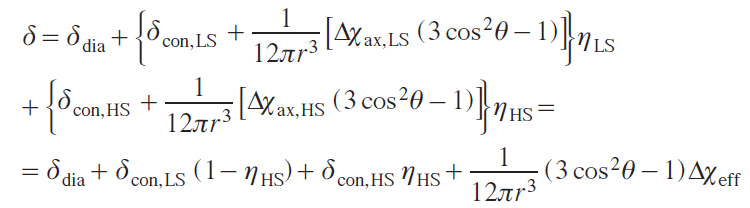
where Δχeff is determined by the following expression:
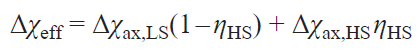
In addition to the chemical shift as a function of the spin state of the complex, another characteristic in the NMR spectrum, viz., the relaxation time, can perform a similar role. From the perspective of the spin dynamics of an NMR experiment, spin equilibrium is a type of chemical equilibrium between two forms: low-spin and high-spin. For each, the position of the signal of the observed nucleus is characterized by a specific chemical shift value. The equation describing the behavior of the nuclear magnetization for such an equilibrium is given by the Bloch – McConnell system of ordinary differential equations.[155] Depending on the ratio of the signal frequency difference to the effective equilibrium constant, either two separate signals (slow exchange) or a single signal, the position of which depends on the equilibrium constant under given conditions (fast exchange), are observed in the NMR spectrum. In the case of fast exchange, the width of the observed signal is expressed by the following equation:[156]
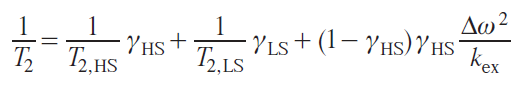
where T2,HS, T2,LS are the transverse relaxation times of the high-spin and low-spin states,gHS is the high-spin fraction sate,kex = kLS → HS + kLS → HS is the exchange frequency between spin states (Hz), Dw is the difference in chemical shifts for two states (Hz).
Approximating experimental nuclear relaxation times within the framework of Eq. (75) allows not only to determine the population of spin states but also to study the kinetics of spin equilibrium. Examples of the application of this approach are described.[147-149]
To conclude, methods have now been developed for studying the spin equilibrium of iron(II), cobalt(II) and iron(III) complexes, as well as its energy and kinetic parameters, using NMR spectroscopy.[157] For other transition metal ions, similar studies are quite rare.
4. Conclusion
NMR spectroscopy of paramagnetic metal complexes is a powerful tool for studying the structure, dynamics, and electronic properties of paramagnetic compounds. Despite significant experimental and theoretical challenges associated with large hyperfine shifts, line broadening, and relaxation effects, modern techniques, including advanced pulse sequences, quantum chemical calculations, and their combination with other spectroscopic methods, have significantly expanded the capabilities of this approach. Effective NMR approaches currently exist for studying spin switches, single molecule magnets, and other magnetic functional compounds. Such studies often rely on quantum chemical calculations, which can contain various uncertainties. Therefore, the development of computational methods is necessary for more effective application of paramagnetic NMR spectroscopy. In addition, NMR studies of the magnetic properties of coordination compounds almost always include the study of the temperature dependence of the observed chemical shifts in the spectra, which requires the appropriate equipment.
Further development of this area of chemistry involves optimizing experimental approaches, improving spectral interpretation, and integrating NMR with other methods, such as EPR and magnetometry. This will allow to gain a deeper understanding of the nature of paramagnetic effects and expand the application of NMR spectroscopy in chemistry, biochemistry, and materials science.
Thus, NMR of paramagnetic complexes continues to be a dynamically developing scientific field, offering unique opportunities for studying the electronic and spatial structure of paramagnetic molecules.
This review was financially supported by the Russian Science Foundation (Project No. 24-73-00257).
5. List of abbreviations and designations
The following abbreviations and notations are used in the review:
AQ — acquisition time,
BIAN — bis(phenylimino)acenaphthenequinone,
CASSCF — complete active space self-consistent field (method),
COSY — correlation spectroscopy,
Cp* — pentamethylcyclopentadienyl,
15C5 — 15-crown-5 ether,
DFT — density functional theory,
d1 — delay between scans in an NMR pulse sequence
DMAO — dimethylalumoxane,
EFG — electric field gradient,
HFC — hyperfine coupling,
HS — high-spin (state),
HSQC — heteronuclear single quantum coherence,
KD — Kramers doublet,
LIS — lanthanide-induced shift,
LS — low-spin (state),
MAO — methylalumoxane,
SMM — single-molecule magnet,
MO — molecular orbital,
MRI — magnetic resonance imaging,
pNMR — paramagnetic NMR spectroscopy,
ppm — parts per million,
SH — spin Hamiltonian,
TEMPO — 2,2,6,6-tetramethylpiperidine 1-oxyl,
THz EPR — terahertz EPR spectroscopy,
Pc — phthalocyanine,
RDC — residual dipolar coupling,
RQC — residual quadrupolar coupling,
TPP — meso-tetraphenylporphyrinate dianion,
ZFS — zero-field splitting,
A — hyperfine coupling tensor,
Aiso — isotropic HFC constant,
Adip — traceless dipolar (isotropic) part of the HFC matrix,
B0 — external magnetic induction,
Biq — crystal field parameters (in the Stevens formalism),
c — concentration of a paramagnetic compound in solution,
Cj — tabular constant for lantanide ions in Bleaney’s theory,
D — zero-field splitting energy (ZFS parameters),
d, d0 — density of the solution with and without a paramagnetic compound,
En — energy of the n-th level (eigenvalue of the spin Hamiltonian),
ΔE — energy difference,
ΔF — free energy difference,
Fk — hyperfine coupling constant for the k-th nucleus,
g — electronic g-factor (tensor),
giso — isotropic value of the g-tensor,
g∥ , g⊥ — longitudinal and transverse components of the g-tensor, respectively,
H→ — magnetic field,¶
DH — spin transition enthalpy,
h — Planck constant,
ħ — reduced Planck constant (ħ = h/2π),
I — nuclear spin (quantum number),
J — quantum number of the total angular momentum of electron (for lanthanides),
kB — Boltzmann constant,
k→ — unit vector in the direction of the external magnetic field,
kex x — exchange frequency between spin states,
L̂ — orbital angular momentum operator,
M — molar weight,
M→ — magnetization,
Mi — magnetization component,
MS — spin magnetic quantum number,
NA — Avogadro’s constant,
Ôiq — Stevens operators,
Pi — population of the i-th energy level,
R — universal gas constant,
Ri — enhancement of nuclear relaxation (longitudinal for i = 1 and transverse for i = 2),
Ricon — contact contribution to relaxation,
Ridip — dipole contribution to relaxation,
RiCurie — contribution of the Curie mechanism to relaxation.,
r — distance,
Rkl — ratio of geometric factors for k and l nuclei,
S — electron spin (quantum number and operator S^S).
⟨SZ⟩ — average electron spin projection,
SLS — order parameter depending on intra-nuclear mobility,
ΔS — entropy of the spin transition,
T — temperature,
T1 , T2 — longitudinal and transverse relaxation times of nuclei,
U — energy barrier to magnetization reversal (for molecular magnets),
γ — gyromagnetic ratio,
γe — gyromagnetic ratio of the electron,
γI — gyromagnetic ratio of a nucleus,
γHS , γLS — fraction of low-spin and high-spin states,
Δ — crystal field parameter,
δ — chemical shift,
δdia — diamagnetic contribution to chemical shift
δcon — contact shift,
δdip — dipolar (pseudocintact) shift,
δpara — total paramagnetic shift,
δij — Kroneker delta,
θ, ϕ — azimuthal and equatorial angles in the spherical coordinate system,
λ — spin-orbital interaction energy (or contribution of chemical bonding to diamagnetic susceptibility),
μ0 — vacuum permeability,
μB — Bohr magneton,
μN — nuclear magneton,
μ — magnetic moment,
η — spin state population,
ν0 — Larmor frequency,
νi — transition frequency of the i-th Kramers doublet,
ρN — spin density at the position of a nucleus,
ρ(r→) — electron density distribution function,
σ — spin – orbit coupling reduction factor,
τ — correlation time,
τr — correlation time of the rotational reorientation of the molecule,
τe1, τe2 — longitudinal and transverse electron relaxation times
τM — residence time,
ticon, tidip, tiCurie — correlation times for the corresponding relaxation mechanisms,
χ — volume magnetic susceptibility,
χ — magnetic susceptibility tensor,
χM — molar magnetic susceptibility,
χMdia — diamagnetic contribution to the molar magnetic susceptibility,
Δχ — anisotropic component of the magnetic susceptibility tensor,
Δχax — axial component of the magnetic susceptibility tensor,
Δχrh — rhombic component of the magnetic susceptibility tensor,
χ– — isotropic (average) magnetic susceptibility,
ωS, ωI — Larmor frequencies of the electron and nucleus.
References












