


Keywords
Abstract
The review addresses potential applications of layered LiNixCoyMnzO2 (x + y + z = 1) oxides in the form of non-agglomerated large-crystalline (singlecrystal) particles as cathode materials for lithium-ion batteries (LIBs). A unique feature of this review is integration of fundamental aspects into a unified material design strategy. Correlations between the thermodynamics of crystal facet surface energy, the chemical potentials of lithium and oxygen, and crystal growth kinetics were established for the first time. This enables targeted control of particle morphology depending on the surface composition and synthesis conditions. The chemistry and thermodynamic conditions of defect formation are described in detail, and existing gaps in their crystallographic description are highlighted. In comparison with other studies, the patterns of mechanical degradation of single crystals during long-term cycling were identified, which made it possible to formulate strategies for effective control of the defect structure to fabricate cathodes with high specific capacity and extended cycle life. Methods for investigating the mechanical properties of particles and methodological limitations of these methods are analyzed for the first time.
The bibliography includes 308 references.
1. Introduction
Rapid climate change and the increasing frequency of natural disasters are increasingly attributed to global warming, which is a result of the industrial era and the growing consumption of fossil fuels. One strategy proposed to mitigate the adverse anthropogenic impact on the environment is the transition from internal combustion engine vehicles to electric vehicles. Despite the ongoing debates about the actual efficiency of this switching for decreasing the CO2 emissions (measured in kg of CO2 per km travelled), which depends on the nature of the electricity source (coal, gas, nuclear power, renewable energy) used to manufacture and charge electric vehicles,[1] these vehicles are being used more and more often as part of global efforts to reduce the carbon footprint. This imposes increasing demands on the next generation of lithium-ion batteries (LIBs), which are currently the most promising rechargeable power source for electric vehicles. Currently, LIBs have achieved specific energies of approximately 300 W h kg–1 at the cell level, which is adequate for electric vehicles with a driving range of up to 600 km.[2][3] However, for vehicles with longer driving ranges, further increase in the energy density of LIBs is required.[4][5]
It is well known that the energy and power characteristics of LIBs are mainly limited by the positive electrode (cathode) material, which is the most expensive component of the battery. Therefore, to overcome the technological restrictions and design a new generation of LIBs with specific energy above 300 Wh kg–1, it is necessary to develop cathode materials with enhanced electrochemical capacity. The layered oxides LiNixMnyCozO2 (x + y + z = 1, х≥0.6, so-called Ni-rich NMC)* are considered to be excellent candidates for modern LIBs owing to their high discharge capacity (≥180 mA h g–1),[6][7] which can be improvedby increasing the nickel content in NMC. However, cathode materials with a high nickel content (x > 0.8), while possessing markedly higher electrochemical capacity, are severely limited in practical applications due to pronounced capacity loss during electrochemical cycling and to low thermal stability, which poses a threat to the safe operation of the battery.[8]
The problem of electrochemical capacity degradation is related, first of all, to microstructural features of cathode materials based on NMC layered oxides. Cathode materials that are currently used in commercial LIBs are mainly based on particles (polycrystalline, PC-NMC, below referred to as polycrystalline NMC) that represent spherical agglomerates of 5 – 15 μm composed of randomly oriented primary particlesof < 0.5 μm (Fig. 1а).[7][9]
![[{"id":"NhNKTB7ZIk","type":"paragraph","data":{"text":"SEM images of PC-NMC (<i>а</i>) and SC-NMC (<i>b</i>) materials. Author’s figure taken from the archive to a publication.<sup>9</sup>"}}]](/storage/images/resized/Q8dPJX8NspY10ZS0jUQvGchZsuZfXYerpBTbSiL7_xl.webp)
The charge/discharge process in NMC includes a few phase transitions accompanied by a sharp decrease in the distance between the transition metal layers in the layered structure.[7][10-12] The sharp change in the interlayer spacing causes deformation of the crystal lattice, giving rise to mechanical stress within the electrode structure and, finally, results in a loss of mechanical integrity (cracking) of the agglomerates consisting of primary grains, thus accelerating the material degradation. These adverse consequences become even more grave in the case of Ni-rich NMC for which more than 80 – 90% of lithium can be extracted during electrochemical cycling. In this case, anisotropic expansion/contraction of the crystallographic unit cell during charge/discharge process results in the formation of microcracks not only at the level of agglomerates, but also at the level of primary particles, which results in a rapiddecrease in the LIB capacity.[13][14]
Apart from the polycrystalline morphology, NMC can be obtained as separate non-agglomerated primary particles of ≥1 µm in size (Fig. 1b) (single-crystal, SC-NMC, or monolithic particles;[15-17] here referred to as single-crystal NMC). However, these particles are not perfect single crystals but rather large crystalline grains devoid of internal grain boundaries characteristic of agglomerated structures. The absence of grain boundaries provides these materials with higher stability during electrochemical cycling due to lower mechanical stress concentrations and a reduced tendency for cracking.[9][18-21] Furthermore, single-crystal NMC of a particular morphology can exhibit a higher tap density than their polycrystalline counterparts; this increases the active material loading per unit area of the electrode, thus increasing the energy density of LIBs, and thereby enhancing safety and mitigating capacity fading owing to the reduced electrode–electrolyte interfacial area.[22-24]
Owing to these advantages, single-crystal Ni-rich NMC are attracting considerable attention among both researchers and industry professionals.[25] Analysis of the trends in the publication activity (Fig. 2) revealed a 7 – 10% annual increase in the number of papers on this subject. This trend is apparently due to both commercial interests of major industry players (such as Tesla Motors[26] and LG Chem[27][28]) and continuing efforts of researchers to improve the performance characteristics of SC-NMC materials.
![[{"id":"fctM3CVyR3","type":"paragraph","data":{"text":"Data on the annual number of publications retrieved by the search for the terms ‘single crystal NMC’ and ‘single crystal NCM’ in Google Scholar over the last seven years from 2020 to 2026. The number of publications was extrapolated on the basis of data for the first four months of 2026."}}]](/storage/images/resized/aK96ILB2nwYUF8kIML672pf40fy15EoYsRzNReee_xl.webp)
Despite the annual increase in the number of original studies and reviews dealing with SC-NMC (see Fig. 2), these studies are mainly focused on comparison of the electrochemical properties of single-crystal and polycrystalline materials and on the search for the optimal method for the synthesis of SC-NMC. The issue of the rational design of the particle morphology for SC-NMC is usually addressed only occasionally in the context of the influence of synthesis temperature or the flux composition in the case where the molten salt (flux growth) method is used for the synthesis. Unlike most review publications available to date,[21][29][30] the present review is focused on analysis of the methods for controlling the morphology of SC-NMC in the context of thermodynamics and kinetics of crystallization processes. Particular attention is devoted to the effect of the SC-NMC morphology on the electrochemical properties, in particular on the bulk lithium diffusion and on the differences between the diffusion characteristics of single-crystal and polycrystalline materials. In addition, the review addresses various types of lattice defects in SC-NMC, the mechanisms of defect formation, and the influence of defects on the mechanical and electrochemical properties and also discusses the strategies for enhancing the electrochemical performance of these materials.
* In the scientific literature dealing with layered oxides based on lithium and transition metals (Ni, Mn, Co), the designations NMC and NCM are used interchangeably, with Ni being traditionally listed first. It is important to note that the formulations with the nickel mole fraction x≥0.6 are classified as nickel-rich ones, whereas materials with x = 0.3 – 0.6 are regarded as containing a moderate proportion of nickel.
2. Morphology control and synthesis of single-crystal NMC
An advantage of SC-NMC is the possibility of rational design of the particle shape and size by using various methods and by varying the synthesis conditions. Experimental studies clearly demonstrate that the SC-NMC morphology is determined by a combination of thermodynamic and kinetic factors that depend on the chosen synthesis method and conditions: temperature, atmosphere, starting reactants, and additives.[31] Thermodynamic and kinetic factors are competing, effective control of particle morphology requires understanding which factor plays a key role in determining the crystal faceting. In the case of thermodynamic control, the shape of the particles formed during the synthesis corresponds to the minimum surface energy (Esurf), whereas in the case of kinetic control, the synthesis proceeds along the path with the lowest energy barrier (Ea) and results in the formation of particles with a greater surface energy (Fig. 3). In the following Sections, the thermodynamic and kinetic factors that determine the SC-NMC morphology are examined in detail.
![[{"id":"tWtq_M45oT","type":"paragraph","data":{"text":"Energy diagram illustrating the choice between thermodynamically (black) and kinetically (red) controlled products. E<sub>a</sub> is the activation energy for the formation of a product of a specified shape, ΔG° is the Gibbs free energy difference between the reactant and the product, Esurf is the surface energy of a product particle. The reactant is a critical-size nucleus and the product is the particle formed upon the synthesis."}}]](/storage/images/resized/tKZGn4GosrFKpdRqA68FBUJvnzxCn5XoXe539xM9_xl.webp)
2.1. Thermodynamic aspects
Ni-rich NMC possesses a layered structure with rhombohedral symmetry (space group R3–m, α-NaFeO2 structural type) composed of two interpenetrating distorted close-packed FCC sublattices, one consisting of oxygen anions and the other containing Li and transition metal (M) cations in alternating (111) planes of the parent FCC structure (Fig. 4).
![[{"id":"EH8HzEe5C4","type":"paragraph","data":{"text":"Crystal structure of a layered lithium and transition metal (M) oxide."}}]](/storage/images/resized/nSDCG8g38IpfogXMACCj54I9mCYpECtNWgIM9YJM_xl.webp)
The equilibrium shape of a particle formed under particular synthesis conditions can be predicted using the Wulff shape construction method.[32][33] This method makes it possible to determine faceting of a crystal with a fixed volume corresponding to the minimum surface energy based on the crystal symmetry and energies of the main crystallographic planes involved in the formation of the particle facets (surface energies). Thus, to determine the equilibrium shape of a particle, it is necessary to know the set of main surfaces and the corresponding surface energies, which are usually calculated using density functional theory (DFT).[34]
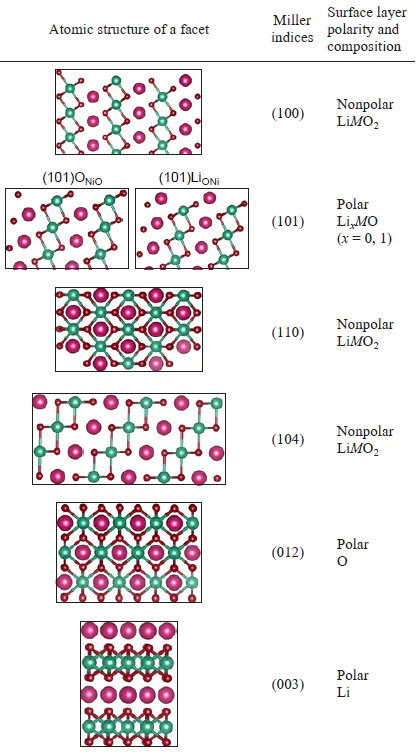
The LiMO2 layered oxides comprise two types of surfaces, nonpolar and polar ones. The nonpolar surfaces are characterized by the Miller indices (104), (110), and (100), while the Miller indices of the polar surfaces are (012), (003), (101), and (111) (Table 1).[35] The surfaces with other Miller indices possess high energy and are not observed experimentally. This subdivision corresponds to the Tasker classification,[36][37] in which the nonpolar surfaces have a zero charge and a zero magnetic moment, while the polar surfaces, in turn, may have a non-zero charge or a non-zero magnetic moment.
The DFT calculations of the surface energy involves the use of special model cells (slabs) containing a bulk phase with two symmetrical surfaces and a vacuum layer (Fig. 5).
![[{"id":"m2VU6Nmo6E","type":"paragraph","data":{"text":"Model slab used to calculate the surface energy."}}]](/storage/images/resized/xAh1Cfysk1F33dt28Thc5gtCe0ZfxBwZRXmAa1MD_xl.webp)
In general terms, the surface energy is defined as the difference between the energy of a slab with two free surfaces and the energy of an equiatomic bulk cell divided by twice the slab surface area A:
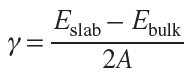
Slabs with symmetric nonpolar surfaces retain their stoichiometric composition, while for polar surfaces, the composition deviates from the stoichiometry. Therefore, the following relation is used to calculate the energy of a polar surface:
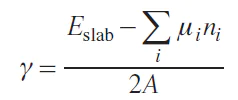
where μi is the chemical potential of an atom of type i, and ni is the number of atoms of type i. The chemical potentials of elements are determined from the thermodynamic equilibrium condition for the particle surface and bulk. For example, the condition of equilibrium for LiNiO2 is written as
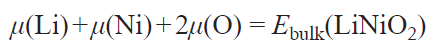
Thus, according to this condition, the energy of a polar surface depends on two independent chemical potentials, μ(Li) and μ(O). Usually, the energy of a lithium metal atom in a BCC lattice is taken as the standard lithium chemical potential μ0(Li), while the standard oxygen chemical potential μ0(O) is the energy of an oxygen molecule with a correction of 1.36 eV, associated with the error of calculation of the molecular binding energy using the PBE exchange-correlation functional.[38] The chemical potentials μ(Li) and μ(O) corresponding to the synthesis conditions are found from the phase diagrams for the Li – M – O systems plotted in the Δμ(Li) and Δμ(O) coordinates using DFT data for the full set of phases in each system (Fig. 6).[37][39][40]
![[{"id":"QGjOcT1lWH","type":"paragraph","data":{"text":"Phase diagrams of the Li – Ni – O (<i>a</i>) and Li – Co – O (<i>b</i>) systems plotted in the Δμ(Li) and Δμ(O) coordinates on the basis of the DFT data. Change in the oxygen chemical potential <i>vs</i>. temperature (<i>c</i>) and partial oxygen pressure (<i>d</i>)."}}]](/storage/images/resized/MzWfaCKYT7683GmTFkNeGdesc4RfsOw86OsMSjek_xl.webp)
The change in the oxygen chemical potential Δμ(O) depends on the temperature T(K) and the partial oxygen pressure p(O2) as
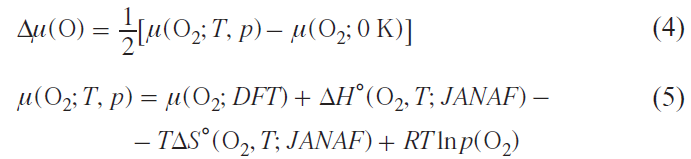
where μ(O2; T, p) is the chemical potential of molecular oxygen, μ(O2; DFT) is the total energy of O2 molecule calculated by DFT, ΔH°(O2 , T; JANAF) is the standard enthalpy of formation of the O2 molecule taken from the Joint Army – Navy–Air Force (JANAF) thermochemical tables,* ΔS°(O2, T; JANAF) is the standard entropy of the O2 molecule taken from the JANAF thermochemical tables, R is the universal gas constant.
The Δμ(O) exhibits an approximately linear inverse dependence on temperature and a logarithmic dependence on partial oxygen pressure (see Fig. 6). Thus, Δμ(O) effectively reflects the synthesis conditions. Meanwhile, Δμ(Li) is definedrelative to the lithium chemical potential metal [Δμ(Li) = μ(Li) – μ(Li metal)] and is often used as a proxy for lithium concentration; however, the quantitative relationship between these two quantities remains unclear.[41]
The chemical potentials of lithium and oxygen affectonly the surface energy of polar surfaces, for LiNiO2 (LNO) and LiCoO2 (LCO) in Fig. 7. Several studies have reported that Co and Mn have only a minor effect on the surface energy of NMC with a nickel content of x ≥0.8. Therefore, the data obtained for LNO can be used to analyze the general surface energy trends in Ni-rich oxide crystals.[35]
![[{"id":"yYs8vAtBfp","type":"paragraph","data":{"text":"Surface energies of LiNiO<sub>2</sub> (<i>a</i>) and LiCoO<sub>2</sub> (<i>b</i>) as functions of the oxygen chemical potential.<sup>40</sup>"}}]](/storage/images/resized/Cqb172nj1w0WLINarVB1cNOMPJoMpzxlh2xh3rAt_xl.webp)
The energy of the considered nonpolar surfaces (100), (110), and (104) does not depend on external conditions due to their stoichiometric composition and zero charge (see Fig. 7). It was found that the (104) surface has the lowest energy and the (100) surface has the highest energy, which correlates with the number of cleaved M – O bonds.[35][42] The second energy minimum corresponds to the (003) polar surface, which forms upon cleavage of the LiMO2 crystal along the lithium layers. In view of the fact that the surface layer represents a lithium monolayer, the energy of the (003) surface is a function of the Li chemical potential. Depending on the conditions, several stable configurations are observed where the fraction of occupied lithium sites in the monolayer ranges from 1/8 to 1 (Fig. 8a,c,e).[29][43-45] For LCO, the stoichiometric composition with a Li occupancy of 1/2 in the surface monolayer ensures the minimum (003) surface energy within the oxygen chemical potential range of −2.6 < Δμ(O) < −1.8 eV (Fig. 8e), whereas for LNO, non-stoichiometric compositions are stabilized over a broad range of oxygen chemical potentials (Fig. 8с). Furthermore, decreasing the lithium chemical potential stabilizes surface configurations with lower Li contents (Fig. 8g).
![[{"id":"lHAeHviAbc","type":"paragraph","data":{"text":"(<i>a</i>) Top view of the (003) surfaces with 1, 1/2, and 0 lithium in the monolayer (ML); (<i>b</i>) top view of the (012) surfaces with 1, 1/2, and 0 oxygen in the monolayer. Energy of the polar surfaces in LNO/LCO as a function of oxygen chemical potential: (<i>c, d</i>) (003) with different lithium contents in the monolayer; (<i>e, f</i>) (012) with different oxygen contents in the monolayer. (<i>g</i>) Energy of the (003) surface with different lithium contents in the monolayer as a function of the lithium chemical potential. The Figure was created by the authors using published data: (<i>c, d</i>),<sup>39</sup> (<i>e, f</i>),<sup>45</sup> (<i>g</i>).<sup>37</sup>"}}]](/storage/images/resized/PxtbCJDKKVCe4M1qaLYjDAssqnOpxfrIilqSGZZ6_xl.webp)
The (012) polar surface exhibits the next lowest energy and, similar to the (104) facet, plays a crucial role in electrochemical processes. This is because lithium diffusion channels terminate at the (104) and (012) surfaces; thus, the contribution of these surfaces to the resulting particle shape in the layered oxides substantially influences the (de)intercalation kinetics. This surface can exhibit a variety of structural configurations depending on the oxygen coverage in the surface monolayer. The stoichiometry is achieved for a composition with 1/2 oxygen monolayer on the surface (Fig. 8b); however, this configuration has a relatively high energy (Fig. 8d, f ). In LCO, the (012) surface energy can be lowered under both the oxidating conditions (increasing monolayer oxygen content) and reducing conditions (decreasing monolayer oxygen content) (Fig. 8f ). In contrast, for LNO, it can be decreased due to the formation of LiNi – NiLi antisite defects (Fig. 8d ).
The calculated surface energies can be used to determine the thermodynamic equilibrium crystal shapes at specified oxygen and lithium chemical potentials. Previous studies have indicated that the Li chemical potential has a lower effect on particle morphology than the oxygen chemical potential.[35][43] However, the chosen chemical potentials must lie within the thermodynamic stability region of LiMO2 in the Δμ(Li) – Δμ(O) phase diagram (see Fig. 6). Consequently, varying the oxygen chemical potential is often coupled with a corresponding change in the lithium chemical potential.[39][40]
The effect of Δμ(O) on the equilibrium crystal shape of the LiMO2 particles is illustrated in Fig. 9. For LNO, the (104) and (003) facets are present over the whole range of Δμ(O), with the (104) facet dominating the morphology. The particle shape is virtually invariant in the 0 > Δμ(O) > −0.93 eV range, which corresponds to the low-temperature synthesis conditions (below 500°C). As Δμ(O) further decreases, the (101) and subsequently (012) regions are formed, due to decreasing contribution of the (104) surface. Consequently, the equilibrium particle shape transforms from a trigonal antiprism formed by the (104) and (003) facets to a truncated cuboctahedron formed by the (104), (003), (012), and (101) planes. Conversely, the particle shape in LCO is determined by the (104), (012), and (003) facets. Decreasing Δμ(O) reduces the contribution of the (003) surface from 55% to 15% and contribution of the (012) surface from 20% to complete disappearance at Δμ(O) = −1 eV. Simultaneously, the percentage of the (104) surface increases from 20% to 85%. As a result, the particle shape changes from a hexagonal prism formed by the (104), (012), and (003) surfaces to an antiprism formed by the (104) and (003) surfaces.
![[{"id":"Tjf1WgQeeR","type":"paragraph","data":{"text":"Variation of the equilibrium shape and contributions of surfaces to the resulting particle shape of LNO, LCO, and NMC811 depending on the oxygen chemical potential.<sup>35, 37, 40</sup>"}}]](/storage/images/resized/t1fxncKu86cVn9a9WB5H6poKXuaS3VVQqFNTTjfe_xl.webp)
For NMC compounds, such as NMC811, the particle morphology is characterized by the presence of (104), (003), and (012) surfaces over the whole considered range of oxygen chemical potentials.[35][43] In an oxygen-rich atmosphere or at low temperatures (0 > Δμ(O) > −1.3 eV), an antiprismatic particle shape is formed predominantly by (104) facets, which is optimal for the extraction of lithium from the layered oxide structure. A decrease in the oxygen potential (corresponding to increasing temperature or conducting the synthesis in an oxygen-deficient atmosphere) results in an increase in the contribution of the (012) facets, as the area of (104) facets decreases until they completely disappear at Δμ(O) = −2 eV (~ 1200°C). Simultaneously, the proportion of (003) facets, which are inaccessible to lithium diffusion, increases twofold, which may potentially deteriorate the power characteristics of the cathode material.
Although theoretical and experimental studies have demonstrated that the Wulff construction based on surface energies reliably predicts the shapes of layered LiMO2 oxide particles, such as NMC333,[29][43] NMC532,[29] NMC622,[29] and NMC811,[29] it is important to note that the real structure of the particle surface may substantially differ from the model structure. For instance, combined theoretical and experimental studies [41][46][47] have shown that doping NMC811 with boron decreases the (003) surface energy relative to the (104) surface energy, thereby stabilizing a plate-like morphology. This energy decrease is attributable to the adsorption of lithium borate, which serves as a source of boron during synthesis; this, in turn, leads to an increase in the lithium chemical potential.[41][46][47] The surface segregation of doping elements can also cause considerable changes in the surface energy, thereby affecting the particle morphology.[48][49] However, it should be noted that that all the trends discussed above apply strictly to thermodynamically equilibrium conditions. The effect of kinetic factors during crystallization on the particle shape is discussed in the next Section.
* NIST-JANAF Thermochemical Tables: official website URL: https://janaf.nist.gov (accessed on June 22, 2026).
2.2. Kinetic aspects
Unlike thermodynamic considerations, crystal nucleation and growth kinetics are influenced by numerous factors acting at different stages of crystallization.. The influence is determined by the balance between the nucleation and particle growth rates, diffusion mechanisms, and synthesisconditions.[50] Therefore, study of the influence of kinetic factors on the particle shape presents a more complex challenge from both experimental and theoretical standpoints.[51-53] Irrespective of the composition, the crystallization of particles follows the patterns predicted by the classical nucleation and growth theory.[54] However, the specific features of SC-NMC synthesis limit the applicability of classical approaches for assessing the influence of kinetic factors on crystal size and morphology. Indeed, as shown below in Section 2.3, the synthesis of SC-NMC may include a step of crystallization from solution resulting in the formation of submicron particles of mixed hydroxide NixMnyCoz(OH)2 or carbonate NixMnyCozCO3 precursors assembled into spherical agglomerates. However, the final size and shape of SC-NMC crystals are determined by the conditions of the high-temperature reaction between the precursor and a source of lithium, in which the morphology of the precursor primary particles and agglomerates is not preserved during lithiation. Currently, there are no comprehensive studies on the influence of kinetic factors on the morphology of SC-NMC crystals. Most likely, Ostwald ripening is likely to be the dominant crystal-growth mechanism, especially if high-temperature lithiation is carried out in the presence of a liquid phase (low-melting-point flux). During crystal growth, redissolution or surface diffusion processes are likely to occur, which is a key factor in the formation of crystal facets. During particle growth, material is deposited relatively uniformly over the crystal surface and can subsequently redistribute via surface diffusion. However, when the rate of surface diffusion is much lower than the particle growth rate, the redistribution cannot keep pace with particle growth, and spherical particles are formed (Fig. 10а).[55]
![[{"id":"Mla_SXlJR8","type":"paragraph","data":{"text":"(<i>a</i>) Schematic picture illustrating the effect of thermodynamics and kinetics on the resulting particle shape; (<i>b – g</i>) effect of the competition between thermodynamic and kinetic factors on the final shape and size of MnCO3 particles; (<i>b – d</i>) SEM images of MnCO<sub>3</sub> particles obtained at initial Mn<sup>2+</sup> concentrations of 1.5, 4, and 12.0 mmol L<sup>–1</sup>, respectively; (<i>e – g</i>) particle shape and size predicted by the computational model: rhombohedral particles formed at the initial Mn<sup>2+</sup> concentration of ∼ 1.5 mmol L<sup>–1</sup> (<i>e</i>), cubic particles formed at the initial Mn<sup>2+</sup> concentration of ∼ 4.5 mmol L<sup>–1</sup> ( <i>f</i>), spherical particles formed at the initial Mn<sup>2+</sup> concentration of ∼ 12.0 mmol L<sup>–1</sup> (<i>g</i>).<sup>55</sup> Copyright American Chemical Society."}}]](/storage/images/resized/TFSDwZGrgcbbg9VM3uON1Yaltk0lm6CIcWr2IMGv_xl.webp)
A comprehensive theoretical and experimental study of the effect of synthesis conditions on the morphology taking into account the kinetic aspects of particle shape evolution was carried out for the model MnCO3 system.[55] Using phase-field simulations, the authors demonstrated that with increasing concentration of metal cations in solution, the high rate of crystal growth promotes a transition from the equilibrium octahedral morphology, first, to a cubic shape and then to a spherical shape, which is accompanied by a decrease in the particle size (Fig. 10). Although the study involved a system containing the salt of only one metal, the authors noted that this approach can be extended to the synthesis of mixed transition metal salts, composition-gradient materials, and other cathode materials.
Thus, the kinetics of crystal growth is determined by a sequence of stages, each being characterized by a particular rate and depending on numerous factors. The first stage involves diffusive transport of the crystallizing species from the bulk medium to the surface of the growing crystal. The driving force for this transport is the chemical-potential difference between the medium and the crystal (μmedium – μcrystal). The diffusion transfer is followed by the adsorption of particles on the crystal surface. The final stage of the process is integration of the adsorbed particles into the bulk crystal. The rate-limiting step in the crystallization kinetics is always the slowest of the above processes. Particular examples of the effect of kinetic factors on the NMC morphology are discussed below, in the context of description of various synthesis methods.
2.3. Synthesis methods
All types of NMC cathode materials, including SC-NMC, must meet a number of technological and performance requirements (Table 2): (1) a homogeneous distribution of transition metals (Ni, Mn, Co) throughout the particle bulk; (2) high tap density; (3) specified particle size distribution; (4) low specific surface area; (5) low moisture content; and (6) moderately alkaline pH of the aqueous extract.[7] As with PC-NMC the chemical composition and spatial distribution of Ni, Mn, and Co are crucial for achieving the target capacity and cycle life in SC-NMC. The tap density, which directly influences the energy density of LIBs, is determined by the particle shape and particle size distribution. This parameter can be optimized through the preparation of particles with appropriate morphology and a narrow size distribution and by using polydisperse systems.
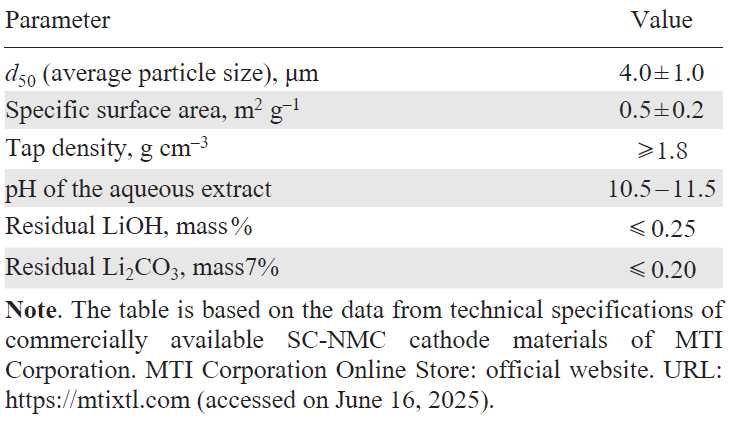
It is known that spherical-like particles[56] provide the highest tap densities owing to their most efficient packing. In the case of spherical particles, the densest packing is achieved in ordered structures such as hexagonal close packing or face-centred cubic lattice in which the space filling factor reaches approximately 74%. However, in real powder systems that were freely poured without external compaction or vibration, monodisperse spherical particles typically form disordered structures with a filling factor of about 64%.[57] Particles with distinct facets, acute corners, and edges tend to form stable arch-like structures that prevent close packing (filling factor of approximately 58 – 60%); therefore, their tap density is lower than that of spherical particles. A low specific surface area is preferred for a cathode material, because this decreases the volume of the electrolyte required for complete wetting of the electrode and also reduces the intensity of the side surface reactions. The pH value of the aqueous extract serves as an indirect indicator of the concentration of surface LiOH and Li2CO3 compounds formed upon contact of the material with atmospheric moisture and CO2 . The solution pH should be moderately alkaline, which makes it possible to avoid the degradation of the polymer binder during the preparation of the electrode slurry (see Table 2).
In order to achieve optimal electrochemical performance, the synthesis of an SC-NMC cathode material should give a material that meets all of the above requirements. Methods for the preparation of SC-NMC can be conventionally divided into two main groups: the precursor-free method and the method involving presynthesized NixMnyCoz(OH)2 precursor. In the latter case, transition metals are typically in the +2 oxidation state (Table 3). However, the oxidation states of cobalt and manganese may differ from the indicated value and depend on the precursor synthesis conditions. In particular, Mn3+/4+ and Co2+/3+ hydroxides can be prepared by varying the atmosphere used for the synthesis and concentration of the transition metal in the reaction medium or by using oxidants (for example, H2O2).[58]
According to the first approach, a transition metal source (e.g., oxide, hydroxide, nitrate, or acetate) is mixed with a source of lithium (e.g., LiOH, LiNO3, Li2CO3), followed by heat treatment.[59-63] Although this approach results in the formation of octahedrally shaped single-phase layered samples (space group R3m), control of the morphology and particle size is virtually impossible in this method.[59][62][63] For the formation of single-crystal particles, not only high temperature (>950°C) should be used, but also long annealing time is required, because diffusion of transition metal ions in oxides is hindered due to the strong M – O bond. If low-melting-point fluxes are used, the long-term heat treatment may be accompanied by Ostwald ripening involving the growth of some particles at the expense of dissolution of other particles. Nevertheless, the particle size usually does not exceed 2 μm.[59]
The synthesis of SC-NMC typically involves a two-stage procedure comprising the preparation of an intermediate (precursor) and the subsequent high-temperature treatment of the precursor as a mixture with a source of lithium. Several methods have been developed for the precursors synthesis, including hydro-[64] and solvothermal methods,[65] sol – gel process,[66] spray drying,[67] spray pyrolysis,[68] and so on. However, co-precipitation is the most common and preferable method for the industrial production of NMC cathode material precursors.[69] In the precursor-based method, the problem of chemical homogeneity is solved by precipitating the precursor with a homogeneous distribution of transition metal cations from soluble salts in the presence of a complexing agent.[70][71] Subsequently, the obtained mixed transition metal precursor is subjected to high-temperature lithiation, as in the synthesis of polycrystalline NMC. This synthetic stage is crucial and particularly this stage determines the morphology of the final product. There are two main protocols used for the lithiation of SC-NMC: one is based on high-temperature (950 – 1050°С) treatment of the precursor mixed with a lithium salt in one or several stages with intermediate grinding of the formed agglomerates,[72-79] while the other protocol uses inert salts or eutectic salt mixtures (flux), apart from the source of lithium, in order to obtain crystals of a cathode material by high-temperature treatment (850 – 950°С) in a melt of these salts (molten salt method).[77][80] Despite the simplicity of the synthesis, the main drawback of the former lithiation method is the use of a higher temperature for solid-state synthesis, which results in higher Ni2+ concentration in the Li sites of the layered structure of Ni-rich NMC and in the formation of disordered phases with a spinel- and/or rock salt-type structure on the particle surface. All the above features have an adverse effect on the electrochemical performance of cathode materials.[81-83] Furthermore, high synthesis temperatures increase the probability of sintering and formation of large agglomerates; therefore, a final milling stage is required to produce cathode material as single-crystal particles. Meanwhile, milling may cause particle cracking or surface cleavage.[84] Moreover, the synthesis conditions do not allow for effective control of the morphology or particle size distribution of the material, since milling process results in the formation of crystals of arbitrary (polygonal) shapes.[74][75][85]
In the molten salt method, a mixture of low-melting salts (e.g., an eutectic mixture) is used as a flux.[58][86-92] The process of SC-NMC synthesis by heat treatment in a flux melt can be divided into three consecutive stages, each characterized by different mechanisms and phase transformations. The first stage involves heating of a reaction mixture consisting of a hydroxide precursor, hydrated lithium hydroxide, and a flux, with all components being in the solid state. This stage is performed at temperatures of up to ~ 200°C where water is removed from lithium hydroxide monohydrate LiOH · H2O, while mixed hydroxide forms a monoclinic MOOH phase (space group C2/m)[93] and lithium-deficient spinel phases upon the reaction with dehydrated lithium hydroxide.[94] In the second stage (200°C ≤ T≤ 500°C), M oxide hydroxides undergo thermal decomposition, while simultaneously reacting with a lithium source to form spinel-like intermediates and lithium-containing compounds of the rock salt structural type (space group Fm3–m).[93] In this case, the process takes place in the solid phase due to the low temperature.[95] The salts used as flux components remain in the solid state because the temperature is not sufficiently high for them to melt. This stage can be described as a pre-lithiation process.[93] The third stage, which occurs at temperatures above 500°C, involves Li/M ordering and the formation of a layered structure. Finally, the intermediate phases are transformed into a layered oxide (space group R3–m) as the temperature rises during the synthesis. The flux components melt, leading to the formation of a liquid phase, accompanied by a dissolution – recrystallization process. The continuous recrystallization process can furnish virtually pore-free particles. Nevertheless, decomposition of precursor particles and the growth of large SC-NMC crystals can produce internal nanovoids.[96][97] The molten salts fill the spaces between the particles, thus effectively increasing the contact area between them.[80] In addition, the diffusion coefficients of transition metal and lithium cations in the melt are much higher than those in the solid phase.[21] When the flux melting point and the temperature of long-term holding (900 – 1000°C) have been reached, the precursor agglomerates rapidly disintegrate, and the stage of growth of single crystallites begins.[80] As the synthesis temperature increases, the molten flux becomes less viscous, which facilitates mass transfer and, consequently, leads to particle growth.[89] An increase in the holding time promotes further disintegration of the initial agglomerates and the continuous growth of single non-agglomerated particles accompanied by the development of their crystal facets (Fig. 11),[80] with the particle growth occurring by the Ostwald ripening mechanism.[98]
![[{"id":"mjIu-FfeA9","type":"paragraph","data":{"text":"SEM images showing the stages of disintegration of precursor agglomerates and the growth of single non-aggregated particles during the molten salt crystallization process as a function of temperature and holding time.<sup>80</sup> Copyright Elsevier."}}]](/storage/images/resized/HPEFjVImgeuplmCnrfxs6QGRIkvQvN0HIYt3P77T_xl.webp)
Differential scanning calorimetry (DSC) was used to identify the key stages of the molten-salt crystallization process occurring in a multicomponent reaction mixture on heating (Fig. 12). Heating of a mixture containing Ni0.8Mn0.1Co0.1(OH)2 , LiOH · H2O, and Li2SO4 (flux) produced a three-stage weight loss profile. The first stage (~ 105°С) corresponds to the elimination of crystallization water from lithium hydroxide. The second stage (~ 280°С) is associated with decomposition of the mixed transition metal hydroxide to the corresponding oxides with the release of H2O.[99-102] The third stage (~ 460°C) is characterized by a gradual weight loss and ends with an endothermic peak associated with melting of residual LiOH. Particularly at this stage, the reaction between the lithium-containing component and the transition metal oxides is initiated, leading to the gradual formation of layered structures.[101-103] At the final stage, cation ordering takes place without any significant change in the weight to give a thermodynamically stable phase corresponding to space group R3̅m.
![[{"id":"4fCcLj_DsD","type":"paragraph","data":{"text":"Thermogravimetric (dashed line) and calorimetric (DSC, solid line) profiles of the reaction mixture consisting of Ni<sub>0.8</sub>Mn<sub>0.1</sub>Co<sub>0.1</sub>(OH)<sub>2</sub>, LiOH · H<sub>2</sub>O, and Li<sub>2</sub>SO<sub>4</sub>."}}]](/storage/images/resized/XjunBL3mxVxYkXcOWv7ZJX0vGOZbcl7Uf67BA3cg_xl.webp)
A key role in the synthesis is played by the concentration of the added salts. A salt deficiency leads to the formation of particles with residual agglomeration and a high proportion of solid – solid interfaces due to insufficient wetting of the particle surface (Fig. 13a,b).[104]
![[{"id":"kkj4y6z7TS","type":"paragraph","data":{"text":"SEM images of SC-NMC111 samples obtained with varying amounts of flux in the synthesis of single crystals: 0 mol.% (<i>a, b</i>), 20 mol.% (<i>c, d</i>), 60 mol.% (<i>e, f</i>), and 80 mol.% (<i>g, h</i>). The high-temperature treatment conditions were the same for all samples.<sup>104</sup> Copyright Royal Society of Chemistry."}}]](/storage/images/resized/BFNl42MRE89qOcojZqRd64dP32Dm3OwXrzVk4zMM_xl.webp)
Conversely, increasing the salt content decreases the number of direct contacts between solid particles, potentially facilitating crystal growth. However, an excess of salt may lead to an increase in the distance between particles, thus retarding the growth (Fig. 13e – h), which is attributable to an insufficient amount of the supplied material due to the need to cover large distances between particles. A moderate excess of inert salts can promote fast growth of NMC crystals (Fig. 13с,d ).[104][105]
Polycrystalline NMC with a relatively high tap density (2.0 – 2.5 g cm–3) are typically synthesized using dense spherical precursor agglomerates with an average size of 10 – 12 μm.[106-108] However, a precursor with a high tap density is unsuitable for the synthesis of SC-NMC, as this leads to residual agglomeration.[95] Therefore, to prepare non-agglomerated, single-crystal particles, it is advisable to use precursor agglomerates no larger than 5 μm with a relatively loose morphology,[95] or non-agglomerated precursor particles.
A major benefit of the molten salt synthesis is the possibility of controlling the SC-NMC morphology by varying the composition of the flux. The variation of the composition of the low-melting-point mixture makes it possible to modify the final crystal shape, which is mainly achieved by altering the lithium chemical potential. Nevertheless, kinetic effects on surface formation cannot be completely disregarded, particularly the influence of melt viscosity, which correlates with the melting temperature of the selected flux. For example, the use of KCl instead of lithium salts decreases the lithium chemical potential and makes it possible to prepare crystals with a cuboctahedral morphology,[92] owing to the reduced contribution of the (003) facet and the enhanced contribution of the (101) facet as well as to elimination of the differences between the surface energies of the (111–) and (003), (012) crystallographic facets.[9] The crystal faceting is defined by the (003) and (101) planes, which are polar (see Table 1) and lithium-terminated, indicating the sensitivity to μ(Li). However, the use of NaCl results in an octahedral morphology due to a higher melting point of the sodium salt compared to the potassium salt and higher viscosity of the melt, which limits mass transfer.[92] It is known that in the presence of Na2SO4 as a flux, the SC-NMC particles acquire an octahedral shape, while the addition of Li2SO4 leads to the formation of truncated octahedra,[89] which is due to lower melting point of Li2SO4 in the presence of LiOH compared to Na2SO4 and to simpler mass transfer.[109][110] Presumably, the interaction of the (003), (101), and (111) facets of the NMC crystals with Na+ ions inhibits the crystal growth in the directions perpendicular to these facets.[89] A CsCl – LiCl mixture (1 : 4) as a flux makes a contribution to the final morphology, giving rise to truncated octahedral particles, because of the use of a small amount of Cs+ salt and more intense growth of the (104) surface. When LiCl is completely removed from the reaction mixture, and only CsCl is used as the molten salt, the growing particles have a polygonal (random) morphology, since μ(Li) decreases, thus increasing the contribution of the (003) surface.[29]
A key objective is to increase the tap density of the cathode powder; this can be achieved by preparing spherical single-crystal particles. For the targeted design of a crystal morphology, relying on the understanding of the kinetic processes occurring during crystal growth by the molten salt method, it is necessary to minimize the contribution of thermodynamic processes that considerably influence the growth of particular facets and to enhance the contribution of the kinetic component. Thus, from a kinetic standpoint, it is necessary to create conditions under which the surface energies of the facets will be equal, in a first approximation. In other words, it is necessary to reduce the surface energy anisotropy of the major crystallographic facets, (104), (003), and (012),[9] which determine the final morphology of SC-NMC. A sharp temperature rise, which enhances the contribution of thermodynamic factors to the final morphology, and tuning of thermodynamic parameters by varying the flux composition would lead to the growth and spread of vicinal surfaces throughout the whole particle surface area and would provide, most likely, spherical and spherical-like particles. Decreasing μ(Li) is favourable for the propagation of the (003) surface, while decreasing p(O2) stabilizes the (104) facet.[29][43]
Thus, a change in the flux composition plays a substantial role regarding both kinetic and thermodynamic parameters. Mass transfer processes may be accelerated upon the introduction of low-melting-point components, which, according to the phase diagrams of the chosen salts and lithium hydroxide, would occur in the liquid phase at the synthesis temperature. Considering thermodynamic factors, at lower chemical potentials of oxygen, e.g., in a weakly oxidative atmosphere or at higher reaction temperature, the (104) facet predominates in the surface over the (012) facet.[29] On the other hand, the (101) and (003) facets are more sensitive to the lithium chemical potential, and their proportion in the surface increases with decreasing μ(Li). The effect of Na+, K+, and Cs+ salts on Li chemical potential and stabilization of the (104), (101), and (100) facets would depend on the chosen flux composition (Table 4)[111][112]; this would produce particles in the form of truncated octahedra, antiprisms, cuboctahedra, hexagonal prisms, and spheres.
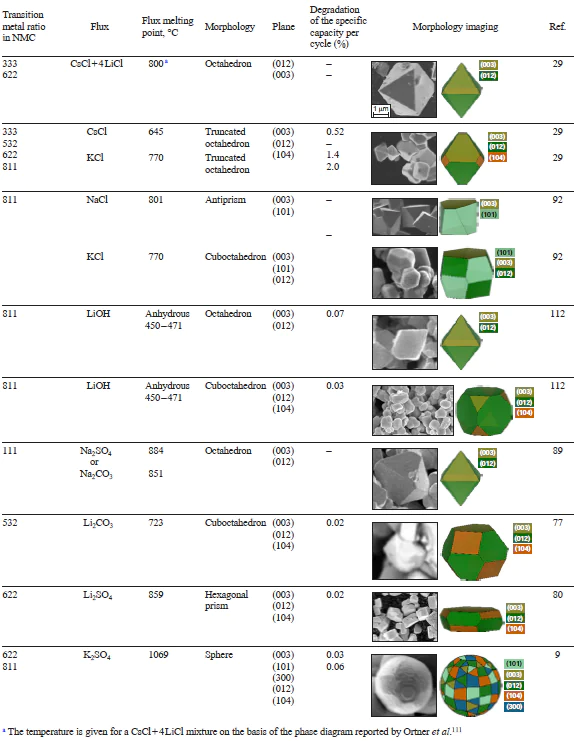
In the case of NMC with a high Ni content, the variation in oxygen partial pressure is markedly limited by the need to suppress the partial occupation of Li sites by Ni2+ cations, which requires an increase in p(O2) to stabilize Ni3+.[113-116] Hence, μ(Li) is still the key parameter to dictate the morphology of Ni-rich SC-NMC crystals. Furthermore, a decrease in the lithium chemical potential promotes the growth of vicinal planes and the formation of spherical-like particles.[9]
Despite the benefits, the molten salt synthesis method has a number of drawbacks. A major problem is that after the synthesis, the cathode material surface bears the flux residues, which are electrochemically inactive and must be removed by washing. However, washing can also make the particle surface more vulnerable to environmental degradation, particularly in the presence of water and CO2 , which can have an adverse effect on the electrochemical properties of the material. This issue is addressed by using resintering, which helps to restore the particle surface and improve the particle stability.
In conclusion, it is noteworthy that precursor-free methods for the synthesis of SC-NMC still represent a poorly investigated area. On the contrary, approaches based on the use of pre-synthesized precursors are well-elaborated and commercially utilized processes that yield SC-NMC with a homogeneous distribution of transition metals among particles, high tap density, and a controlled particle size distribution. Meanwhile, solid-state methods have a number of limitations (see Table 3), despite their simplicity and extensive use. The limitations include high (950 – 1050°C) temperatures of the synthesis, long annealing times, the need for intermediate milling and/or lithiation steps, as well as limited control over particle morphology, which mainly includes only controlled particle size. The molten salt synthesis also provides control over the shape of SC-NMC particles via variation of the chemical potentials of lithium and oxygen. However, since this synthesis method includes several heat treatment steps and washing to remove flux components and residual lithium compounds, it remains a more labour-intensive, time-consuming, and energy-intensive process than conventional solid-state synthesis.[30][83][117]
3. Electrochemical properties
To date, the electrochemical properties of SC-NMC have been investigated most extensively for particles with octahedral, truncated octahedral, or polygonal (irregular) morphologies. Among these, polygonal particles predominate owing to the widespread use of the solid-state synthesis followed by mechanical crushing or milling of the cathode material particles. However, establishing quantitative relationships between the electrochemical performance of SC-NMC and particle morphology (e.g., Savina and Abakumov[118]) remains challenging because of the limited amount of available statistical data. This is primarily due to the fact that most studies have focused on comparisons between SC-NMC and PC-NMC rather than on systematic investigations of morphology-dependent properties within SC-NMC materials. Generally, SC-NMC materials exhibit specific discharge capacities comparable to those of PC-NMC at low cycling rates, whereas the cycle life and rate capability of single-crystal particles are superior to those of their polycrystalline counterparts (Values describing the degradation of specific discharge capacity per cycle are discussed in Sections 2.3, 4.1.3, and 4.1.4). To understand the origin of these differences in the electrochemical behaviour between SC-NMC and PC-NMC, it is necessary to consider the diffusion and kinetic characteristics of the charge/discharge processes in NMC-based cathode materials.
3.1. Bulk lithium diffusion
Layered oxides are characterized by two-dimensional lithium diffusion within (003) type planes, while diffusion along the [001] direction is possible only in the presence of vacancies or LiM defects in the M layers (Fig. 14).[119][120]
![[{"id":"xHYy9rQJj0","type":"paragraph","data":{"text":"Lithium diffusion in layered oxides"}}]](/storage/images/resized/CILAvmaYoTEqvtTWB8osTdKDvtznTzFIOvB7GaPa_xl.webp)
The key mechanisms of lithium diffusion in layered oxides are vacancy[119] and divacancy[121][122] mechanisms (Fig. 15а). The former involves movement of Li+ between the octahedral LiO6 sites along the shortest path through oxygen dumbbells.[119][123] The latter includes movement along a curved path running through intermediate tetrahedral voids (Fig. 15а).[119][123] Although the energy barrier for the divacancy mechanism is half that of the vacancy one, this mechanism becomes accessible only in the presence of lithium divacancies. In the early stages of charging of layered oxides, the divacancy concentration is generally insufficient to make a significant contribution to the diffusion. Therefore, in these stages, the vacancy mechanism remains the dominant lithium transport mechanism. The divacancy mechanism starts to play a noticeable role when the concentration of lithium vacancies exceeds ~ 0.2 (Fig. 15а).[121][124] Indeed, delithiation is accompanied by an increase in the lithium diffusion coefficient, due to increasing concentration of lithium vacancies; this results in a higher contribution of divacancy diffusion mechanism, which has a lower activation energy.[81][125]
![[{"id":"T6HGlAylyp","type":"paragraph","data":{"text":"(<i>a</i>) Lithium-ion diffusion coefficient in the course of NMC delithiation. The background colours (green and pink) illustrate the predominant area of vacancy (oxygen dumbbell hopping, ODH) and divacancy (tetrahedral site hopping, TSH) mechanisms of lithium transport, respectively; (<i>b</i>) general view of non-equivalent paths (1 – 6) of lithium migration by the vacancy mechanism within the (003) plane, depending on the local environment and the corresponding activation energies; (<i>c</i>) calculated and experimental values of lithium diffusion coefficients in NMC with different Ni, Mn, and Co ratios; (<i>d</i>) lithium diffusion coefficients as a function of voltage at 20°C for LNO samples synthesised with a Li/Ni ratio ranging from 0.90 to 1.02. The Figures were created by the authors of the review using published data: (<i>a – c</i>),<sup>119</sup><sup>, 121</sup> <i>d</i><sup>129</sup>."}}]](/storage/images/resized/idCkjCl3kQwGIsOtGSfUSPsBjkU7AZtA1NzIqcqW_xl.webp)
In view of the fact that the migration barriers for lithium ions strongly depend on the М cations surrounding the diffusion pathway, the lithium diffusion depends on the composition of SC-NMC (Fig. 15b).[119][121] According to DFT calculations, the presence of nickel in the second coordination sphere of the migrating lithium cation results in a reduction of the migration barriers for both the vacancy and divacancy mechanisms (Fig. 15b). The oxidation state of nickel also plays a role: the migration barriers for the divacancy mechanism with neighbouring Ni2+ and Ni3+ cations are 0.36 and 0.43 eV, respectively. The presence of cobalt or manganese increases in the lithium diffusion barriers for both mechanisms.[113][121] Although faster lithium diffusion is expected for compositions with higher nickel contents, in some experimental and computational studies, the highest lithium diffusion coefficient was found for NMC622 (Fig. 15c, Table 5). This is due to the largest interlayer spacing in this material among the NMC compositions considered, which increases the size of the diffusion channels and decreasing barriers for lithium migration.[121][126]
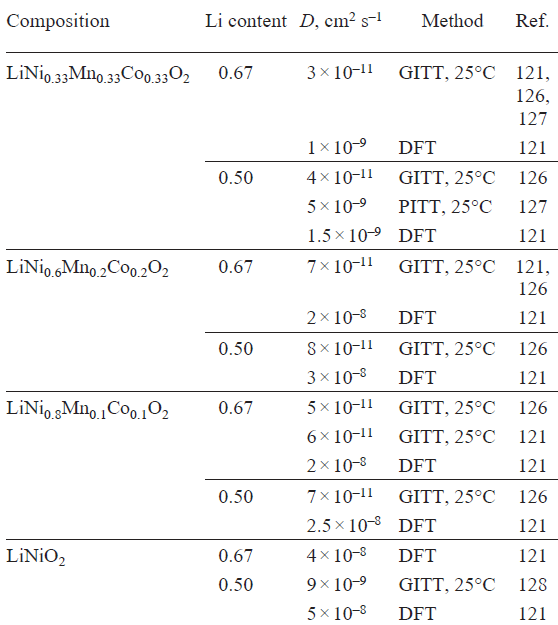
It should be noted that experimentally determined lithium diffusion coefficients strongly depend on the measurement technique employed. For example, the values obtained using galvanostatic intermittent titration technique (GITT) and those determined by the potentiostatic intermittent titration technique (PITT) differ by approximately two orders of magnitude (see Table 5)[127][128]. Furthermore, lithium diffusion is affected by a variety of factors, including external conditions, particle size, and defect concentration. For example, in the case of LNO, it was shown[129] that increasing the concentration of NiLi defects by 4% leads to an approximately one-order of magnitude decrease in the lithium diffusion coefficient (Fig. 15d ).
3.2. Features of lithium diffusion in SC-NMC and PC-NMC
Polycrystalline NMC materials contain primary crystallites measuring 100 – 500 nm, which form micron-sized agglomerates; this facilitates the migration of lithium from the centre of primary particles to the surface, as the small size of crystallites shortens the diffusion path. As a result, the vacancy diffusion mechanism, which predominates in the early stages, is rapidly replaced by a divacancy mechanism, which minimizes kinetic hindrances within the agglomerate bulk (Fig. 16)[130]. Thus, the lithium transport in PC-NMC is more efficient and the effect of diffusion barriers decreases.
![[{"id":"Ro9aRBZdLI","type":"paragraph","data":{"text":"<i>Operando</i> transmission X-ray microscopy and X-ray absorption near-edge structure images of single-crystal and polycrystalline NMC particles demonstrating variation of Ni oxidation state during charging. (<i>a – i</i>) Representative maps of nickel oxidation states taken every 40 min during charging up to 4.4 V at 0.15 C rate.<sup>130</sup> Copyright Wiley."}}]](/storage/images/resized/4sg9KFcH4b6uwpeqD8rSGA7UBNCrdUa7RvuPWRSC_xl.webp)
Numerical modelling of lithium transport in polycrystalline materials, using LCO as a model system, demonstrates that the primary microstructural features governing Li diffusion are the size and misorientation of the primary crystallites.[131] In the case of small-size crystallites (~ 0.2 – 0.4 μm), a crucial factor is the contribution of grain boundaries, whereas in the case of larger crystals (~ 1.6 μm), relative misorientation is the primary factor. Despite the shortening of the lithium diffusion path within the small crystallites in PC-NMC, the presence of grain boundaries has an adverse effect on lithium diffusion.9, 112, 120 The boundaries between primary crystals that are randomly oriented relative to one another act as barriers to lithium migration, thus hindering the diffusion path. In addition, grain boundaries accumulate local mechanical stresses during electrochemical cycling; this results in the formation of microcracks and destruction of agglomerates.[132] Electrolyte penetration into the cracks increases the active surface area of the material; this may temporarily accelerate lithium diffusion owing to exposure of new regions for (de)intercalation. However, this phenomenon is detrimental for cycle life of the cathode material, as grain boundary degradation compromises the mechanical integrity of the agglomerates, while an increase in the surface area in contact with the electrolyte accelerates surface degradation.
The larger particle size of SC-NMC compared with the primary crystallites of PC-NMC results in slower lithium diffusion (Fig. 16, single crystal); this is especially evident for high degree of charge.[133][134] Within a single crystal, diffusion predominantly occurs via the vacancy mechanism, which is characterized by a relatively low transport rate due to the limited number of lithium vacancies. In contrast, on the particle surface where the concentration of lithium vacancies is markedly higher, the divacancy mechanism providing faster lithium migration predominates. As the degree of delithiation increases, the difference between the concentrations of lithium vacancies on the surface and in the bulk of SC-NMC becomes more pronounced, and the limited diffusion in the bulk is the key retarding factor.[135] These diffusion limitations in SC-NMC lead to a lithium concentration gradient, which is the main cause for mechanical stresses and the formation of intragranular cracks.[136-138] Furthermore, the absence of grain boundaries in SC-NMC eliminates problems associated with intergranular diffusion.[120][139] Several studies have reported that SC-NMC show higher power characteristics compared to PC-NMC at various current densities.[80][112][132][140-144] There are data indicating that faceting of SC-NMC crystals also affects their power characteristics. For example, polygonal crystals of SC-NMC811 with predominating (014) surfaces have a higher discharge capacity at high cycling rates (6 С) and a higher diffusion rate of lithium compared with octahedral SC-NMC811 crystals in which the predominating surfaces are (012).[112][137] However, the underlying reasons for this behaviour have not yet been elucidated.
4. Defects in the layered structure
The close-packed crystal structure of layered LiMO2 oxides (see Fig. 4) tends to form various types of defects, which have a substantial effect on the structure stability and electrochemical properties. In the subsequent Sections, we successively consider the main types of defects, their origin, features of formation, and the effect on the SC-NMC structure evolution and degradation during electrochemical cycling.
4.1. Point defects
Point defects include lithium, oxygen, and transition metal vacancies, antisite defects, small polarons, as well as defect complexes representing various combinations of the above types.[49][145-148] Most studies have focused on oxygen vacancies and antisite defects (MLi , LiM), the special case of which are antisite defect pairs (MLi – LiM) (Fig. 17). These defects have been described in detail in a number of studies,[40][145][148-150] including studies addressing nickel-rich NMC.[116][151]
![[{"id":"serSEzdVHY","type":"paragraph","data":{"text":"Point defects of a layered structure in relation to LNO (ideal case): (<i>a</i>) oxygen vacancy, (<i>b</i>) antisite defect pair, (<i>c</i>) NiLi defect, (<i>d</i>) LiNi defect."}}]](/storage/images/resized/DoJbCvzYRiGKRMdz2c5r7yD4sqHBMwHIfeeXYamF_xl.webp)
In this study, we present a comparative analysis of point defect formation in SC-NMC and PC-NMC samples of identical stoichiometry, and examine the effect of morphology on the defect structure of the material.
Although the types of point defects present in PC- and SC-NMC are similar, their spatial distribution and thermodynamic stability differ significantly.[152][153] In polycrystalline materials, defects predominantly form along grain boundaries and interfaces, where the local partial oxygen pressure and the chemical potentials of the constituent elements vary during synthesis.[154][155] Conversely, in single-crystal materials, defects are distributed more uniformly throughout the bulk due to the absence of grain boundaries and more intensive homogenization that occurs during the molten salt synthesis, owing to the presence of the liquid phase. As a result, the probability of aggregation of point defects is lower in SC-NMC than in PC-NMC, which reduces the risk of microcracking but may also limit the rate of lithium diffusion.[152] Thus, differences in the morphology determine the type of defect formation and the subsequent structural evolution of the material. Evidently, higher synthesis temperatures used for single-crystal SC-NMC particles (~900°C) compared with PC-NMC (≤750°C) also influence the thermodynamics of defect formation.
Due to the absence of direct literature data dealing with comparative analysis of defect formation in PC-NMC and SC-NMC structures, the subsequent analysis is based on a comparison of scattered experimental and theoretical data, while the drawn conclusions should be regarded as our working hypothesis.
4.1.1. Oxygen vacancies
Oxygen vacancies are often regarded as a highly important type of point defects in layered oxides, which can considerably influence their thermodynamic stability, electronic structure, and capacity degradation during electrochemical cycling.[83][156][157] However, the extent to which these defects are actually present in layered oxides remains a matter of debate.
Experimental data show contradictory results. The Rietveld structure refinement based on X-ray powder diffraction data sometimes reveals oxygen non-stoichiometry associated with the presence of oxygen vacancies;[158-160] however, this method may underestimate the actual oxygen content and thus create the illusion of oxygen vacancies.[161] More sensitive techniques for determining oxygen site occupancy, such as neutron diffraction and NMR spectroscopy[162][163] have revealed that the bulk of layered oxides is free of oxygen vacancies under both high states of charge (e.g., in charged LCO[161][164] and NaCoO2)[165][166] and high-temperature synthesis conditions, as demonstrated for LNO.[167]
In terms of charge balance, the formation of an oxygen vacancy leads to the release of two electrons, which reduce the transition metal cations in the structure (Fig. 17а).[49][168][169] In the Kröger – Vink notation, this reaction in relation to LNO can be written as
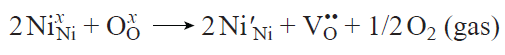
The electrons released upon the oxygen loss reduce the Ni3+ ions to Ni2+ ions.
According to DFT calculations, the formation energy of oxygen vacancies on the surface of layered oxides decreases in the charged state and becomes negative at high states of charge, whereas the vacancy formation energy in the bulk remains positive, thus making the vacancies thermodynamically unfavourable (Fig. 18).[49] [169] This is in line with experimental data, indicating that oxygen vacancies predominantly arise on the particle surface,[158][170] from where they can diffuse to the bulk and induce the formation of stacking faults, micropores, and regions with a rock-salt structure (space group Fm3– m).[158][170-172] However, despite the possibility of vacancy migration to the interior, the concentration of vacancies in the particle bulk should not become high, since the formation and stability of such vacancies are limited by the thermodynamic factors indicated above.
![[{"id":"_2GU80YQBI","type":"paragraph","data":{"text":"Effect of the partial oxygen pressure (<i>a</i>) and temperature (<i>b</i>) on the formation energy of an oxygen vacancy in the bulk of LiхNiO2 (х = 0, 1) and on the (104) surface of Li<sub>x</sub>NiO<sub>2</sub> (х = 0, 1).<sup>169</sup>"}}]](/storage/images/resized/yomoyhl52vVrmKqi53GQAEuAgMmomTTzBjQfctYt_xl.webp)
The particle morphology has a considerable effect on the thermodynamics of oxygen vacancies: oxygen loss is predominantly initiated along grain boundaries and in surface regions; therefore, oxygen loss is more pronounced in polycrystalline NMC than in SC-NMC due to the different numbers of active sites.[150][154][169][173-176] This is confirmed by experimental data, which show that the amount of gas, including oxygen, released from the electrochemical cell during charge/discharge process is much lower for SC-NMC than for PC-NMC, even at elevated cycling potentials.[154]
Conversely, calculations indicate that the oxygen vacancy formation energy decreases with increasing temperature both at the surface and in the bulk.[169][173](Fig. 18b). However, a neutron diffraction study did not reveal oxygen vacancies in LNO synthesized at temperatures below 750°C.[167] This discrepancy can be explained as follows: despite the fact that the formation of vacancies becomes thermodynamically more favourable at high temperatures, their formation in the crystal bulk is highly limited by high energy barriers for migration (1.2 – 1.8 eV in the bulk vs. 0.2 – 0.3 eV on the surface[177]) and by the additional barrier for the formation of dimers with O – O covalent bond (≈ 2.5 eV[173]). Thus, even at high temperatures, oxygen stoichiometry is apparently maintained within the crystal bulk due to the high energy required for the formation and migration of oxygen vacancies.
Crucially, oxygen non-stoichiometry observed in the synthesis or electrochemical cycling[82][178][179] does not necessarily indicate the formation of oxygen vacancies. The structural changes such as transitions from a layered structure to a spinel-like structure, then to a disordered layered structure, and finally to a rock-salt-type structure[133][180][181] may be accompanied by increasing occupancy of the octahedral sites with the М cations, which decreases the О/M ratio, while full occupancy of oxygen sites is maintained.
To compensate for oxygen loss and achieve the target stoichiometry of NMC during high-temperature synthesis, two complementary strategies are commonly employed: (i) using a lithium excess in the precursor mixture, and (ii) annealing in an oxygen-rich atmosphere. However, the latter approach is mainly relevant for nickel-rich NMC.[9][113][114] [182][183] The oxygen loss from the surface regions of the particles can be mitigated by deposition of protective coatings[184][185] or by doping with elements prone to surface segregation.[49][186]
4.1.2. Antisite defect pairs
An antisite defect pair is often regarded as a model defect, since the energy required for its formation serves as an indicator of the proneness of layered oxides to disorder.[40] This type of defect arises when lithium and transition metal ions exchange positions, leading to the formation of a pair of antisite defects LiM and MLi (Fig. 17b). Thus, the local chemical composition of the material remains unchanged, but charge redistribution may take place. Indeed, Ni3+ in the lithium site is reduced to Ni2+; in order to maintain the charge balance, one of the adjacent Ni3+ ions should be oxidized to Ni4+.40 In the Kröger – Vink notation, this reaction can be written as
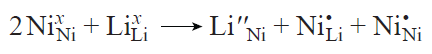
In terms of thermodynamics, the energy of formation of a pair of antisite defects is determined by the difference between the total energies of the defective and ideal structures. Among the transition metals in the series LiNiO2 – LiCoO2 – LiMnO2 , the lowest antisite pair formation energy is inherent in nickel: E(Ni3+Li – LiNi) = 0.62 eV, which is markedly lower than that for Co3+ (1.80 eV) or Mn3+ (1.96 eV).[40][187] However, some of nickel in NMC occurs in the Ni2+ state.7 In this case, the formation of the Ni2+Li – LiNi antisite pair is not accompanied by the formation of an additional Ni4+ polaron and requires even less energy. Indeed, a systematic study of the effect of the composition of layered oxides on the formation of antisite pairs demonstrated that upon the addition of manganese to LNO, the Ni2+Li – LiNi antisite formation energy decreases almost to zero (Fig. 19а).[178] This is attributable to the tendency of Ni2+ to occupy lithium positions, especially in nickel-rich NMC in which cation mixing easily occurs due to similar ionic radii of Ni2+ (0.69 Å) and Li+ (0.76 Å).[116] However, the dimensional factor alone fails to explain the observed dependence of the degree of Ni/Li disorder on the composition. In particular, an increase in the fraction of Ni3+ in nickel-rich NMC is accompanied by enhancement of the cation mixing despite the fact that the Ni2+ concentration simultaneously decreases.[188][189] This indicates that, along with geometric factors, electronic and magnetic effects play an important role.
![[{"id":"rXILKajpX5","type":"paragraph","data":{"text":"(<i>a</i>) Effect of the composition of layered oxide on the formation energy of antisite (AS) pair. (<i>b</i>) <sup>190</sup> Schematic view of superexchange interactions in the NiLi – O – M complexes."}}]](/storage/images/resized/jpibtRWvN820cG0KDnbTVKw38rlAcCkJblBPVWTS_xl.webp)
According to published data,[190][191] a key factor for stabilization of antisite pairs is superexchange interaction between transition metal ions occurring through the oxygen anions (Fig. 19b). The insertion of Ni2+ into lithium sites leads to the formation of energetically favourable Ni2+ – O2− – Ni2+ antiferromagnetic interactions, which markedly reduce the energy required for the formation of antisite defect pairs. In nickel-rich NMC, Ni3+ is indirectly involved in the defect formation, since after occupying the lithium position, it is reduced to Ni2+ and is included in the superexchange interactions. Thus, the high degree of Ni/Li disorder in nickel-rich NMC is not so much caused by the absolute concentration of Ni2+, but by the possibility of local formation of Ni2+ and subsequent defect stabilization through magnetic interactions. The roles of Mn and Co in this process differ significantly. Manganese (Mn4+) promotes the formation of Ni2+ in the transition metal layer and, therefore, enhances the superexchange interactions through the formation of strong Ni2+ – O2− – Ni2+ bonds, thus decreasing the energy of antisite pair formation. Conversely, the low-spin Co3+ ion is non-magnetic and is not involved in the superexchange; hence, it weakens the magnetic interactions between nickel ions and thus suppresses the tendency for NiLi – LiNi antisite pair formation. However, the exchange event is followed by local charge compensation: the neighbouring Co3+ is oxidized to Co4+. This Co4+ ion can form new superexchange bonds, Ni2+ – O2− – Co4+, thus additionally stabilizing the NiLi – LiNi defect, and may promote further Ni/Li exchange.
In summary, antisite defect pairs provide a simplified model of cation disorder in layered oxides, illustrating the interplay between composition, magnetic interactions, and the stability of the layered structure. However, cation exchange in real layered oxide crystals is rarely a strictly reciprocal process. Depending on the synthesis conditions and the state of charge, asymmetric defects may form during electrochemical cycling, whereby one type of ion occupies crystallographic sites that are not native to it without a corresponding migration of the counter-cation. Such defects contribute to the evolution of cation disorder and may significantly influence the structural stability and electrochemical performance of the material.
4.1.3. LiM substitution defects
Substitution defects in which lithium enters a transition metal site (LiNi, Fig. 17c) are relatively rare, although they can occur in lithium-rich systems (Li1 + xM1 – xO2) during the synthesis at moderate temperatures (700 – 800°C) where the mobility of transition metal cations is low[179][191] or in the presence of a large excess of LiOH · H2O in the high-temperature synthesis (up to 50 mol.% in the case of SC-NMC vs. < 5 mol.% for PC-NMC).[9][80][176][182][183][192][193] According to the Kröger – Vink notation, this process can be represented in the following way:
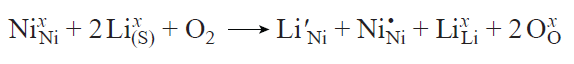
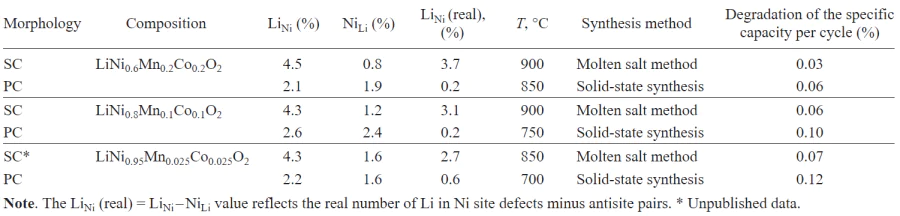
Table 6 presents a comparison of experimental data on the concentration of LiM defects in SC-NMC synthesized by the molten salt method with an excess of lithium hydroxide with analogous data for PC-NMC prepared by solid-state synthesis. SC-NMC has a higher concentration of LiNi defects in which lithium occupies nickel sites. This is due to the use of n (precursor) : n (LiOH · H2O) molar ratio of 1 : 1.5 in the synthesis of SC-NMC, which is much higher than the excess of lithium hydroxide used for the synthesis of PC-NMC (< 5 mol.%). For estimation of the proportion of LiNi defects, it is necessary to take into account the contribution of antisite pairs by applying an appropriate correction to find the true concentration of the substitution defects. The corrected values are summarized in Table 6[194].
4.1.4. NiLi substitution defects
The substitution defects associated with insertion of nickel into lithium sites (NiLi) are the most characteristic and thermodynamically favourable type of point defects in nickel-rich NMC. They are formed both during the synthesis and during electrochemical cycling, when local Ni3+ → Ni2+ reduction takes place and Ni2+ cations migrate to the lithium sites.[116][190] According to the Kröger – Vink notation, this process can be represented in the following way:
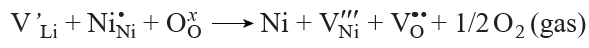
In LiNixMnyCozO2, an increase in the nickel content is accompanied by a decrease in the concentration of the highly charged Mn4+ cations; hence, charge compensation is required to maintain electrical neutrality. This compensation is achieved by the partial oxidation of Ni2+ to Ni3+. When the partial oxygen pressure decreases, for example, if the synthesis is carried out in an air rather than in an oxygen atmosphere, oxygen is released from the crystal lattice to the gas phase. As a result, Ni3+ is reduced to Ni2+, which tends to migrate to lithium sites to give NiLi type defects. According to calculations, the presence of oxygen vacancies markedly lowers the barrier for nickel migration into the lithium sites (Fig. 20а,b).[173] Simultaneously, nickel and oxygen vacancies annihilate, which triggers a structural transformation: transition from the layered LiMO2 structure to a rock-salt structure (Fig. 20с).[81] As noted above, oxygen vacancies are mainly generated on the surface of particles and along grain boundaries; therefore, structural rearrangement is often observed particularly in these parts of the crystals.
![[{"id":"UNLu3nUXjw","type":"paragraph","data":{"text":"(<i>a</i>) Diagram of nickel migration into the lithium layer in the presence of an oxygen vacancy (V<sub>x</sub>O) at one of three considered sites. (<i>b</i>)<sup>173</sup> Migration barriers for nickel to the lithium site calculated using the elastic band method in an ideal structure and in the presence of an oxygen vacancy at the sites shown in (<i>a</i>). (<i>c</i>) Illustration of the degradation of the layered NMC structure to a rock salt structure. The pink circles/polyhedra show sites occupied with lithium, while the green ones indicate sites occupied with transition metals. The smaller red circles designate the oxygen sites."}}]](/storage/images/resized/DlKRPsxrZBqL1mUOFtdQU3kjFbMPviuZXFy6QSy7_xl.webp)
The concentrations of NiLi in NMC as functions of the morphology (SC/PC), composition, and conditions of synthesis are analyzed in Table 7[195-209]. When solid-state synthesis is used, the fraction of NiLi tends to decrease with increasing synthesis temperature for both polycrystalline and single-crystal NMC. However, in the case of single-crystal NMC811 synthesised by the molten salt method the density of defects is markedly lower than that for SC-NMC811 samples prepared by the solid-state method at a similar temperature. According to the data of Table 7, an analogous trend holds for the composition of NMC622 single crystals. This feature can be attributed to kinetic limitations of the solid-state synthesis. Despite thorough mixing of the starting components, the relatively slow lithium diffusion ions in the solid phase leads to local lithium deficiency in particular regions of the growing crystal, especially in the interior of the crystal, where Li+ access is limited. In these lithium-deficient regions, nickel ions, first of all Ni2+, thermodynamically tend to occupy lithium sites, thus forming NiLi defects.
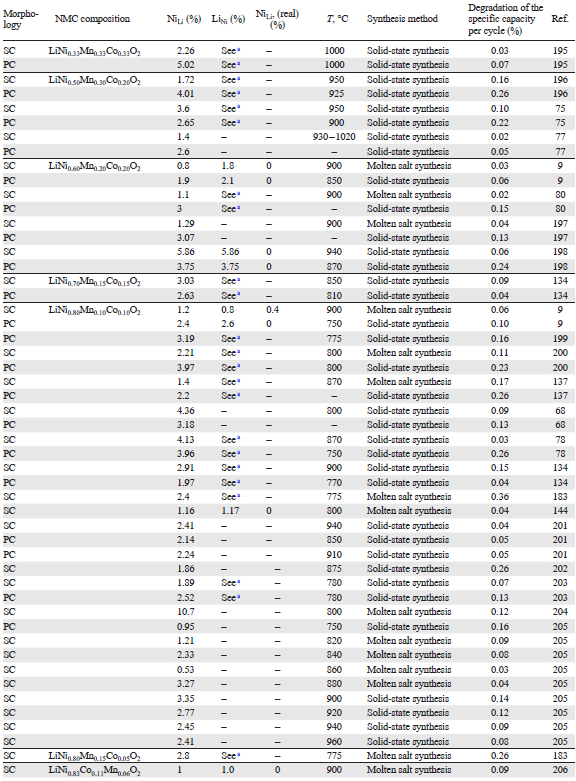
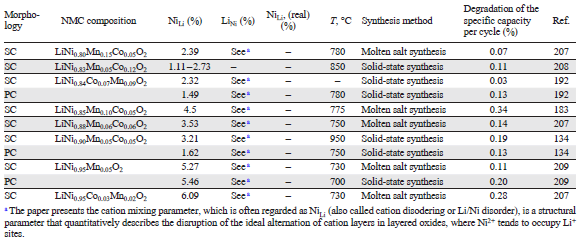
In contrast, the molten salt method minimizes the probability of formation of lithium-deficient regions, owing to the liquid reaction medium, which facilitates lithium transport into the hydroxide precursor structure, which prevents the formation of regions with elevated concentrations of Ni2+.[210][211] The key factor is the use of an excess of lithium source (LiOH · H2O), some of which is spent for the lithiation of the hydroxide precursor, while the rest serves for the formation of the melt. As a result, a high lithium chemical potential is maintained in the system; this creates conditions for the reverse process: the insertion of lithium ions into nickel sites accompanied by the oxidation of Ni2+ to Ni3+. As a consequence, the formation of NiLi defects is strongly suppressed (see Table 7). The molten salt method is, therefore, considered to be preferable over solid-state synthesis for the preparation of single-crystal particles with a reduced number of NiLi defects.
However, it is important to note that the data presented in Table 7 cannot be regarded as entirely objective. This is due to the lack of information on the proportion of antisite defect pairs or the concentration of LiNi defects. In addition, it was found[212] that the duration of the high-temperature treatment has a pronounced effect on the concentration of antisite defects in the solid-state synthesis. Thus, an increase in the holding time promotes more complete homogenization of lithium and transition metals in the crystal lattice, which, in turn, results in decreasing concentration of NiLi type defects (Fig. 21). Therefore, despite the large body of available data, a direct comparison of reported defect concentrations remains challenging because of the simultaneous influence of multiple synthesis- and composition-related factors.
![[{"id":"un4cnWy1eU","type":"paragraph","data":{"text":"Effect of the high-temperature annealing time on the occupancy of lithium sites (<i>3b</i>) by nickel in NMC.<sup>212</sup>"}}]](/storage/images/resized/9wlOD8xNqcceD8jnWNgX1ImhMZ4BDGWVOEaerQSa_xl.webp)
4.2. Dislocations and stacking faults
During electrochemical cycling, layered lithium transition-metal oxides undergo a sequence of structural phase transformations, involving a transition from the initial trigonal H1 phase through the monoclinic M phase to the hexagonal H2 and H3 phases. Characteristic features of the changes in the crystal and electronic structures of nickel-rich oxides upon the phase transitions have been considered in detail in a recent review.[7] The H2 ↔ H3 transition is considered to be most destabilizing, as it is accompanied by a sharp change in the interlayer spacing along the c axis of the unit cell. This results in the development of considerable mechanical stresses, which initiate a rearrangement of the oxygen sublattice and the formation of stacking faults as local disruptions in the ideal sequence of oxygen layer packing.[12][213-215] At the atomic level, this process involves the formation of stacking faults due to layer gliding and, consequently, the cubic close-packed O3 structure of oxygen layers (ABCABC sequence) is transformed into the hexagonal close-packed O1 structure (ABAB sequence).[216-218]
The degree of delithiation plays an important role in the formation of stacking faults. Using DFT, it was shown[219] that the formation energy of stacking faults markedly depends on the lithium content in the structure. In partially lithiated states (Li content > 50%), the energy of stacking faults amounts to hundreds of mJ m−2, which makes their formation energetically unfavourable. However, in a highly charged state, this energy substantially decreases to approach zero or even become negative, thus creating a thermodynamic driving force for the formation of O1 layers in the original O3 structure.
It is believed that gliding of layers and the formation of stacking faults are caused by the movement of dislocations, that is, linear defects of the crystal lattice.[220][221] The main gliding plane for both edge and screw dislocations in NMC is the basal (003) plane.[206] However, there are also publications that describe cases of edge dislocations with Burgers vectors being directed away from the basal plane, for example:
● b = 1/3[11̅1] — gliding is accompanied by a stacking disruption in the anionic sublattice, a sharp decrease in the c parameter, and the formation of nanocracks;[222]
● b = 1/6[211] — results in the formation of antiphase boundaries, which impede the lithium diffusion;[223]
● b = 1/2[001] — arises in the case of surface generation of dislocations.[224]
It should be noted that the moiré contrast observed in transmission electron microscopy images is often interpreted as evidence of dislocations or stacking faults.[225][226] However, moiré fringes may also arise solely from the interference of crystal lattices belonging to domains with a slight relative rotation or small differences in lattice parameters, even in the absence of actual linear defects. Therefore, interpretation of such contrast features requires caution and, whenever possible, should be supported by complementary characterization techniques.
4.3. Microcracks in the structure caused by defect formation
Crystal structure defects influence the electrochemical behaviour of cathode materials. In particular, the accumulation of defects within the crystal increases internal mechanical stresses. The relaxation of these stresses occurs through the initiation and propagation of microcracks, which finally cause degradation of the positive electrode and decrease the cycle life. The understanding of the crack formation mechanisms is essential for the reasonable choice of strategies for stabilization of single-crystal materials and suppression of degradation processes.
Presumably, cracking in SC-NMC is related to the formation and movement of edge dislocations caused by the loss of cathode material during electrochemical cycling.[222] According to a different model,[227] cracks in the primary crystallites are caused by gliding of close-packed layers relative to one another along the (003) planes. Since this gliding is reversible only partly, it causes mechanical stresses and layer separation. Microcracks in SC-NMC appear as a result of a multistage mechanism initiated by so-called layer-by-layer delithiation under high stress, resulting in anisotropic change in the crystal structure along the c axis and the subsequent H2 ↔ H3 phase transition.[228] The change does not occur simultaneously throughout the crystal, but propagates gradually, thus generating local shear stress at the interface between the changed and unchanged parts of the crystal.[136] In order to mitigate this stress, gliding occurs in the crystal along the (003) planes. In the early stages, especially at a moderate voltage (up to ~ 4.3 V), this process is reversible: upon the discharge, the layers return to their original positions and no microcracks appear.[136] However, for higher degrees of delithiation (> 4.4 V) or on long-term cycling, the stress is accumulated irreversibly, and microcracks of gliding planes arise as a collapsing interface between the adjacent M-layers (Fig. 22a).[206][227] These cracks are not repaired during discharge, because they are accompanied by lattice collapse.
![[{"id":"5aiW_9futX","type":"paragraph","data":{"text":"(<i>a</i>) Diagram illustrating the formation of cracks due to plane gliding and corresponding images obtained using high-resolution scanning transmission electron microscopy (STEM).<sup>227</sup> Copyright Elsevier. (<i>b</i>) Diagram illustrating the formation of cracks due to the lithium concentration gradient and high-resolution STEM images in the vicinity of the cracks.<sup>138</sup> Copyright American Chemical Society. HAADF-STEM is the high-angle annular dark field scanning transmission electron microscopy, ABF-STEM is the annular bright field scanning transmission electron microscopy."}}]](/storage/images/resized/CQsveS6N9FlS4OZZrrJsnSeiUJCL4D5WC5E4Xqy5_xl.webp)
Microcracks may form due to the development of lithium concentration gradients in localized regions (Fig. 22b) at high current densities.[138] During effective extraction, lithium is primarily removed from the surface region of the particle, while the lithium concentration in the bulk remains relatively high. The arising sharp lithium gradient[229] induces non-uniform anisotropic deformation of the crystal lattice: surface layers are compressed along the c axis to a much greater extent than the inner layers.[229-231] This induces a distribution of internal mechanical stress, that is, the particle surface experiences tensile stress, while the particle core is compressed.[138] In addition, local inhomogeneity in the lithium distribution may be accompanied by Li+/Ni2+ cationic disorder and the formation of domains with a rock-salt structure. These defects act as an additional source of stress and serve as nucleation sites for microcracks.[232][233] During long-term cycling in the fast-charging mode, the accumulation of this stress initiates the formation of microcracks arranged perpendicular to the c axis (along the ab plane).
Several studies have hypothesized that the mechanism of microcrack initiation[234] involves the formation of nano-sized pores within the crystal lattice. These defects can arise both during the high-temperature synthesis of cathode materials[155] and during long-term electrochemical cycling.[234-238] However, it should be noted that the amount of experimental data on single-crystal particles with nano-sized voids available to date remains insufficient to unambiguously establish the mechanism of crack formation.
Currently, the mechanisms of structural degradation leading to the formation of microcracks have not been adequately studied. Single-crystal particles represent a convenient model system for elucidating the nature of these processes, as they make it possible to study in detail the types of cracks and conditions of crack formation. During degradation, two main types of microcracks can be distinguished: surface cracks and bulk cracks (see Fig. 22). However, systematic research is still required to establish the quantitative criteria of crack formation, elucidate the predominant development mechanisms, and determine the relationship between the material composition, morphology, and susceptibility to cracking.
5. Mechanical properties
The electrochemical degradation of Ni-rich NMC cathode materials is primarily driven by their mechanical properties. During repeated lithium (de)intercalation, a number of irreversible mechanical processes develop in both PC-NMC and SC-NMC structures. These processes include intergranular cracking characteristic of polycrystalline materials,[233][239] and intragranular cracking of crystallites in the case of single-crystal particles.[240][241] The appearance of these defects indicates that the mechanical stress arising in the crystal lattice during cycling exceeds the critical values required for crack initiation and activation of gliding systems.
Owing to the absence of grain boundaries and agglomeration, single-crystal particles generally have greater mechanical stability than the polycrystalline analogues. This is favourable for preserving particle integrity over all stages ranging from manufacture of electrodes to operation of an electrochemical cell. Establishing a relationship between the mechanical stability (mechanical characteristics) and degradation processes (particle cracking) during cycling is an important scientific and applied task aimed at the development of materials with a predictable service life and enhanced resistance to cyclic loading.
The key mechanical parameters used to characterize the NMC materials include the elastic modulus (Young’s modulus, E), hardness (H), and fracture toughness (critical stress intensity factor, K1C). These characteristics are typically determined using the nanoindentation method with indenters of various geometries, which are selected depending on the goal of a particular study. The Berkovich indenter, a truncated diamond pyramid, is used most often to determine E and H values by the Oliver – Pharr method.[242] The compression tests using a modified Berkovich indenter provide additional insight into the mechanisms of elastic-plastic deformation and failure.[243] The fracture toughness is determined using trigonal pyramidal indenters with a cube-corner geometry (the angle between the pyramid faces and the vertical center line is 35°)[244] or the standard Berkovich indenter either by direct measurement of the crack length or by analysis of extra penetration (pop-in) effect in the indentation curves.[245]
Among the existing approaches to mechanical testing of NMC cathode materials (indentation of single particles,[246][247] densely sintered particles,[248] etc.), the method based on testing of the micropillars cut out from grains with a specified crystallographic orientation is considered to be the most reliable.[249] This approach minimizes the contribution of intergranular effects and agglomeration, thus providing data that most closely reflect the true mechanical properties of the material.[250]
Using various nanoindentation methods, the elastic modulus and hardness were determined for SC-NMC materials. The results confirm the higher mechanical stability of SC-NMC compared to polycrystalline NMC. For the latter, the Young’s modulus usually varies in the range of 100 – 200 GPa, with the hardness being 6 – 15 GPa.[251] Numerous studies have shown that the electrolyte does not have any considerable effect on the mechanical properties of NMC, as evidenced by the comparability of the E, H, and K1C values measured in both dry and wet states.[252][253] Table 9[254-256] presents the elastic modulus and hardness values for compositionally different SC-NMC materials under various loads.
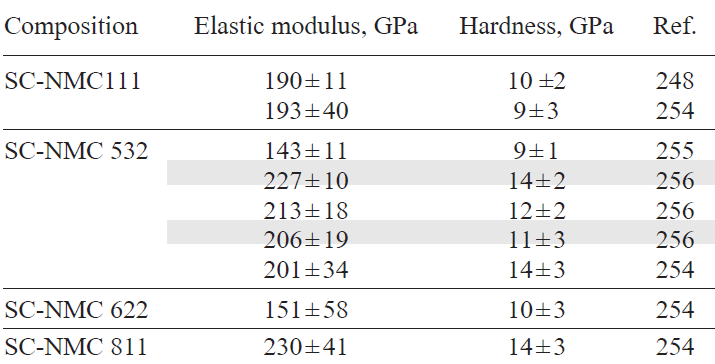
The effects of the degree of delithiation and the number of cycles on the micromechanical properties have been studied in detail for various PC-NMC.[253][255][257] In all cases, the elastic modulus, hardness, and fracture toughness were found to decrease, especially during the first charge/discharge cycle. This behaviour is mainly due to the accumulation of microcracks along the grain boundaries of secondary agglomerates.[239][258] Additionally, it was shown that the strength of polycrystalline agglomerates increases as their size decreases and drops sharply after just a few charge/discharge cycles.[259] Thus, measurements carried out for polycrystalline samples largely characterize the mechanical response of secondary agglomerates and grain boundaries rather than the intrinsic properties of separate crystallites of the active material. Therefore, these results cannot be directly extrapolated to single-crystal particles, in which the effects of intergranular agglomeration and internal grain boundaries are virtually absent. In this context, study of single-crystal particles is of particular interest for determination of intrinsic mechanical properties of the material.[259]
It was shown that delithiation markedly decreases the strength of SC-NMC: the total strength of SC-NMC811 decreases from ~ 86 MPa in the fully lithiated state to ~ 40 MPa in the charged state (Fig. 23d ).[260] For this low strength, plastic deformation and crystal fracture may occur during cycling. Unlike polycrystalline materials, the fracture of SC-NMC involves not only microcrack growth but also cleavage along the basal plane, indicating pronounced mechanical anisotropy inherent in the layered crystal structure.
![[{"id":"qZ-PFph5zR","type":"paragraph","data":{"text":"Post-deformation SEM images of micropillars of the NMC811 single-crystals showing the activation of different slip systems (shown for hexagonal prisms): (<i>а</i>) basal, (<i>b</i>) prismatic, (<i>c</i>) pyramidal; the dot and the arrow point to the slip plane and direction, respectively. In all SEM images, the scale bar is 1 μm. (<i>d</i>) Ultimate strength depending on the lithiation degree of NMC. (<i>e</i>) Map of elastic moduli calculated along all crystallographic planes of NMC using stiffness constants. (<i>a, b, c</i>) <sup>249</sup> Copyright Elsevier, (<i>d</i>) <sup>260</sup> Copyright IOP, (<i>e</i>) <sup>254</sup> Copyright Elsevier."}}]](/storage/images/resized/YwUJOQX3AsCrGIm9XSw2qH6kDPtkMCIfFrrSQpzK_xl.webp)
The anisotropy of mechanical properties was confirmed both experimentally and theoretically. The indentation results showed that the highest elastic modulus corresponds to the (001) basal plane, whereas for the other prismatic planes (100), (110), and (210), the elastic modulus is much lower (Fig. 23e). Wang et al.[249] investigated slip systems and critical shear stresses needed for their activation by compression of micropillars cut out from grains with known crystallographic orientation on a sintered NMC811 pellet. The basal {0001}⟨1120⟩ (Fig. 23a), prismatic {101̅0}⟨112̅0⟩ (Fig. 23b), and pyramidal {1̅012}⟨112̅0⟩ (Fig. 23c) slip systems were activated, with the corresponding critical shear stresses being 1.7, 2.3, and 2.4 GPa, respectively.
Notably, most studies of the mechanical properties of single-crystal NMC have been conducted for densely sintered pellets containing large grains resulting from high-temperature sintering. This approach simplifies sample preparation, but does not fully reflect the conditions of actual electrochemical cycling, which is accompanied by repeated reversible changes in the unit cell volume. In addition, traditional nanoindentation methods generate mainly compressive stress, whereas the formation of microcracks and shear deformations in SC-NMC are initiated by tensile stresses.[231] While interpreting published data, it is highly important to take these factors into account.
Generally, high values of the elastic modulus, hardness, and strength play a key role in providing the stability of SC-NMC during long-term cycling. Stability to cyclic mechanical loads is directly related to suppression of microcrack formation and, as a consequence, to increasing cycle life of the cathode material. In this regard, further development of approaches directed towards enhancement of the mechanical and electrochemical properties of single-crystal NMC is a relevant task.
6. Strategies for improving the properties of single-crystal NMC particles
The absence of grain boundaries, enhanced mechanical stability, and low specific surface area of SC-NMC have a favourable effect on their cycle life compared to that of polycrystalline materials. Nevertheless, even for SC-NMC, some of the degradation processes including the growth of microcracks, parasitic cathode–electrolyte reactions, cation mixing, and phase transitions are still relevant. These phenomena are particularly pronounced when the cathode material is operated at elevated voltages (above 4.4 V vs. Li/Li+). Currently, the most common strategies for mitigating degradation processes are doping and the deposition of protective coatings.
The SC-NMC modification by doping with Mg2+ cations is widely addressed in the literature.[94][182][261][262] The high interest is due to the fact that Mg2+ ions can occupy both octahedral М sites and lithium sites due to the similarity of the ionic radii of Li+ (rLi+ = 0.76 Å) and Mg2+ (rMg2+ = 0.72 Å).[263] Depending on the composition of NMC and the state of charge, the presence of Mg2+ ions in the lithium layer induces so-called pillar effect for NMCXYZ (x≥0.8) or the rivet effect for NMCXYZ (x ≤0.8). In particular, the pillar effect is most pronounced at a high degree of delithiation (≥ 4.3 V vs. Li+/Li) of the material near the H2 ↔ H3 phase transition[182] in which the Mg2+ cations located at the octahedral sites in the Li layer act as structural pillars that restrict the contraction of the interlayer spacing. This decreases the anisotropic change in the unit cell volume and, hence, reduces the tendency for cracking[182][263] and simultaneously suppresses the М/Li cation mixing.[264][265] Meanwhile, when the degree of delithiation is moderate (≤4.3 V vs. Li+/Li), the rivet effect operates; therefore, gliding of close-packed layers and the O3/O1 transition are inhibited, and the tensile stress and cracking of cathode particles are also mitigated.[182]
Doping with Al3+ cations effectively inhibits the Li/Ni disorder and the H2 ↔ H3 phase transition, thus providing a ~ 7 – 9% gain in the cycle life.[265-269] Dopants based on Zr4+,[270] Ti4+,[271] and Ta5+ (Refs [272] and [273]) are also widely used for modification of single-crystal NMC, resulting in longer cycle life.[274][275]
Co-doping strategies such as Al/Zr,[276] Zr/Ti,[277] and Ti/Mg/Al[186] are of particular interest. They provide a combined effect, including increased structural stability, improved power characteristics as a result of decreasing energy barriers for Li+ migration, and enhanced chemical stability of the material to the action of electrolyte.
A new promising approach to the surface modification of single-crystal NMC is doping with elements prone to surface segregation, that is, preferential enrichment of the dopant in the near-surface region relative to the bulk. Examples of these elements are Zr,[278] Al,[279] Sb,[280] and Ti.[49][281] Currently, this strategy is among the least studied; however, in recent years, the interest in the surface modification through segregation has been steadily increasing, as evidenced by the publication of both theoretical and experimental studies aimed at identifying dopants with a pronounced tendency toward segregation and elucidating the mechanisms by which they affect surface properties of cathode particles. It was shown that surface-segregating elements can stabilize the surface structure, decrease the surface reactivity toward the electrolyte, and suppress the interfacial parasitic reactions. Altogether, this retards the surface degradation and increases the cycle life of single-crystal cathode materials.[280][282][283]
An approach alternative to doping is application of protective surface coatings to single-crystal NMC particles. Protective coatings can be deposited by atomic layer deposition (ALD),[284] chemical vapour deposition (CVD),[285] or wet chemistry methods.[286][287] The ALD and CVD deposition techniques give uniform coatings with a controlled thickness; however, their use is limited by their high cost and engineering complexity.[288] The wet chemistry methods are more facile and cost-effective, but generally they do not result in the formation of uniform layers of dopants on the particle surface.[289]
A variety of materials have been used as effective protective coatings to decrease the contribution of parasitic side reactions between single-crystal NMC cathode materials and the electrolyte, e.g., oxides MgO,[290][291] TiO2,[287][292][293] MoO3,[284] ZrO2,[294] and Al2O3,[285][295][296] ionic conductors such as LiAlO2,[297][298] Li2ZrO3,[299] LiV2O4,[300] and lithium phosphate Li3PO4.[286]
Regardless of the coating composition, degradation is generally suppressed, primarily due to the inhibition of parasitic cathode – electrolyte interfacial reactions, particularly at elevated temperatures.[301] If compounds used as coatings possess good electronic or ionic conductivity (Al2O3,[285] [295][296] Li2ZrO3,[299] LiV2O4,[300] etc.), enhancement of power characteristics of SC-NMC is additionally noted.[284][292-294][296][298][300]
Thus, the choice of a modification strategy for SC-NMC cathode material is dictated by the specific conditions of operation: the need to operate at high voltages (above 4.3 V), use high cycling rates, or the requirement to maintain the specific discharge capacity relative to the initial value upon long-term electrochemical cycling. Analysis of the available literature indicates that, with respect to cyclic stability, doping typically increases cycle life by 7 – 12% relative to unmodified materials,[182][263][266-273] whereas surface coatings provide improvements of approximately 5 – 9%.[287] [290][293][294][296][299] The combination of bulk doping and surface coating strategies appears particularly promising, as it may provide synergistic improvements in both cycling stability and rate capability of single-crystal NMC cathodes.
7. Conclusion
An increase in the nickel content of cathode materials based on NMC layered oxides makes it possible to achieve a specific capacity of > 180 – 200 mA h g–1, which is necessary for developing LIBs with high energy density. However, electrochemical cycling causes an abrupt change in the unit cell volume of these materials, which generates mechanical stresses in the electrode structure and, finally, results in a loss of mechanical integrity (cracking) of the particles, thus accelerating the material degradation. The effect of these changes on the stability of a material considerably depends on the material morphology. In the case of PC-NMC, in which the primary grains are randomly oriented, repeated changes in the unit cell volume during charge/discharge not only induce intergranular cracking, but also disrupt the integrity of the primary grains; this occurs under the influence of stresses generated by neighbouring particles. When microcracks reach the surface, new surfaces are exposed for contact with the electrolyte, which penetrates deep into the particles, leading to their further degradation. Conversely, in SC-NMC, deformation occurs virtually in the absence of external mechanical stress, because the influence of neighbouring particles is absent. This morphology has a higher stability during long-term electrochemical cycling, which makes single-crystal NMC particularly attractive for the development of high-performance cathode materials. Nevertheless, SC-NMC also exhibits intrinsic limitations associated with slower Li+ diffusion, which are generally less critical in polycrystalline counterparts. In particular, diffusion limitations can lead to the development of Li+ concentration gradients within crystallites during (de)lithiation, which is regarded as one of the major challenges for SC-NMC cathodes. These factors may decrease the power performance of cathode materials based on SC-NMC and cause the accumulation of internal stresses due to anisotropic changes in the unit cell parameters in single particles. Meanwhile, there are data indicating that, depending on the crystal faceting, SC-NMC can demonstrate higher power performance than PC-NMC at high cycling rates. However, no systematic studies along this line are available so far. Fundamental mechanisms of crack formation, which directly determine the cyclic stability of cathode materials, are also poorly addressed in the literature. Despite experimental evidence for the existence of two types of cracks, surface cracks and bulk cracks, there are no systematic descriptions of the nature of their differences, electrochemical conditions of formation, or the kinetics of their subsequent development. Addressing these knowledge gaps is essential for the rational design of strategies aimed at improving the long-term stability of single-crystal cathode materials.
One of the key advantages of SC-NMC is the ability to precisely control particle morphology through the use of low-melting inert salts (fluxes) or eutectic flux mixtures during the synthesis, which opens up extensive opportunities for the rational design of a large range of cathode materials with enhanced electrochemical characteristics. In particular, a key advantage of spherical or spherical-like single-crystal particles is their high tap density, which increases the specific loading of the active material on the electrode and, as a result, provides a high volumetric energy density of LIBs based on such materials. Such precise morphological control enables tailoring cathode properties to specific application requirements while simultaneously enhancing mechanical robustness. However, although the morphological design of PC-NMC is addressed in quite a few studies, analogous works dealing with SC-NMC are still rare. Extending microstructural design concepts developed for PC-NMC to single-crystal materials may provide performance gains beyond those achievable through conventional strategies such as bulk doping or surface coating. Meanwhile, not all strategies for improving the functional properties developed for polycrystalline NMC can be directly applied to single-crystal materials. In particular, the synthesis of structures with a gradient composition or a core–shell structures is difficult. This is due to the fundamentally different synthesis conditions of PC-NMC and SC-NMC materials: whereas nickel-rich polycrystalline materials are usually prepared at temperatures below ~ 750°C, the formation of single-crystal particles requires temperatures of approximately 900°C or higher (Section 1.3).[9][41][77][78][80][116][194][196-199] In addition, the most favourable method for the synthesis of SC-NMC is the molten salt (flux growth) method in which high temperatures and the presence of a viscous liquid medium promote the intense diffusion of transition metals and, as a result, homogenization throughout the whole bulk of the crystal.
Single-crystal NMC with low and moderate nickel content have already proved to serve as stable cathode materials due to less pronounced changes in the unit cell volume during long-term electrochemical cycling. However, Ni-rich SC-NMC are still not widely used as LIB electrodes. Accordingly, the development of synthesis routes for Ni-rich single-crystal materials with enhanced cycling stability remains an important research priority. In particular, single-crystal LiNiO2 is of particular research interest, as it has the highest theoretical specific capacity among layered oxides. Nevertheless, in practice, it is rarely possible to achieve both high specific capacity and satisfactory stability during long-term cycling.[302-305]
Another promising trend is the use of SC-NMC as high-voltage cathode materials (4.8 V vs. Li+/Li).[29][76][306] Another potential application of SC-NMC is that in all-solid-state batteries,[307][308] which make it possible to avoid the use of liquid electrolytes that promote the degradation of electrodes. The key goals are to ensure the compatibility of the SC-NMC surface with a solid electrolyte, optimize the interface between these materials, achieve the close contact between the positive electrode and the solid electrolyte, study the electrochemical behaviour of the cathode material, and develop effective methods for the assembly, including preparation of electrodes. Overcoming these challenges could pave the way for the development of high-energy-density, long-lived, and safer next-generation batteries.
Finally, SC-NMC and PC-NMC are increasingly being viewed as complementary rather than competing cathode materials. Owing to differences in their particle-size distributions, mixtures of SC-NMC and PC-NMC offer opportunities for designing densely packed electrodes with enhanced volumetric energy density. For example, smaller single-crystal particles can efficiently fill the voids between larger polycrystalline agglomerates, leading to improved electrode packing. Such hybrid cathode architectures may combine the superior cycling stability of SC-NMC with the favourable rate capability of PC-NMC, thereby delivering both high power performance and long-term durability.
The review was written with the financial support of the Russian Science Foundation (Project No. 23-73-30003, Sections 4 – 7; and Project No. 23-73-01258, Sections 2 – 3).
8. List of abbreviations and symbols
D — diffusion coefficient of lithium,
d50 — average particle size,
E — elastic/Young modulus,
Ea — activation energy of the product formation,
Esurf — surface energy,
H — hardness,
K1C — critical stress intensity factor (measure of fracture toughness),
LiM — interstitial defect of lithium in the transition metal site,
М — transition metal,
MLi — interstitial defect of transition metal in the lithium site,
MLi – LiM — antisite defect consisting of a transition metal ion in the lithium site and a lithium ion in the transition-metal site,
ni — number of i type atoms,
ΔG° — Gibbs free energy difference between the reactant and the product,
ΔH° (O2) — standard enthalpy of formation of an O2 molecule,
ΔS° (O2) — standard entropy of O2 ,
μi — chemical potential of i-type atom,
μ(Li) — lithium chemical potential,
μ(O) — oxygen chemical potential,
O1 — hexagonal close-packed structure of oxygen layers, ABAB sequence,
O3 — cubic close-packed structure of oxygen layers, ABCABC sequence,
p(O2) — partial oxygen pressure,
R — universal gas constant,
T — temperature,
VxO — oxygen vacancy,
ABF-STEM — annular bright-field scanning transmission electron microscopy,
ALD — atomic layer deposition,
BCC — body-centred cubic lattice,
CVD — chemical vapour deposition,
DFT — density functional theory,
DSC — differential scanning calorimetry,
FCC — face-centred cubic lattice,
GITT — galvanostatic intermittent titration technique,
HAADF-STEM — high-angle annular dark-field scanning transmission electron microscopy,
LCO — LiCoO2,
LIB — lithium-ion battery,
LNO — LiNiO2 ,
ML — monolayer,
NMC — LiNixMnyCozO2 (x + y + z = 1),
NMC333 — LiNi0.33Mn0.33Co0.33O2 ,
NMC532 — LiNi0.5Mn0.3Co0.2O2 ,
NMC622 — LiNi0.6Mn0.2Co0.2O2 ,
NMC811 — LiNi0.8Mn0.1Co0.1O2 ,
ODH — oxygen dumbbell hopping,
PC-NMC — polycrystalline NMC,
PITT–potentiostatic intermittent titration technique,
SC-NMC–single crystal NMC,
SEM–scanning electronic microscopy,
STEM — scanning transmission electron microscopy,
TSH — tetrahedral site hopping,
References




















