


Keywords
Abstract
Quantifying fundamental concepts of chemical bonding, such as ionicity, covalency, effective atomic charges, and the decomposition of cohesive energy into chemically interpretable contributions, remains a persistent challenge in theoretical chemistry. This work reviews a recently developed first-principles methodology based on Wannier functions possessing atomic orbital symmetry, which provides a rigorous framework for numerically characterizing these bonding attributes across diverse systems. We survey the method’s theoretical foundations and application to a wide range of materials, highlighting key results that validate its reliability and universality. Examples demonstrate how the approach captures essential physics of chemical bonding, and bridges conceptual models with quantitative analysis. By synthesizing the accumulated evidence, this review underscores the method’s utility as a robust tool for bonding analysis and discusses its limitations and future prospects.
The bibliography includes 19 references.
1. Introduction
At the most fundamental level, all chemical systems are governed by quantum mechanics, with chemical bonding arising from the interplay of electronic exchange and correlation effects — phenomena that are inherently electrostatic in nature. While a complete quantum-mechanical treatment of electrons and nuclei provides a rigorous theoretical foundation, such an approach offers limited intuitive insight, particularly for complex systems like molecules and solids. A more pragmatic and widely adopted framework simplifies this complexity by focusing on atomic entities and their bonds, categorizing interactions into distinct yet often overlapping types: ionic bonding, characterized by electron transfer and long-range electrostatic forces between charged species; covalent bonding, marked by localized electron density accumulation between atoms and short-range directional interactions; metallic bonding, an extreme case of delocalized multicenter covalent interactions with nearly uniform valence electron density; van der Waals interactions, weak forces arising from fluctuating atomic or molecular multipoles. These bonding types never exist in isolation. For instance, van der Waals forces are ubiquitous in all chemical systems, while even highly ionic systems exhibit some degree of covalency. This simplified classification proved remarkably powerful, enabling researchers to rationalize a vast array of chemical and physical phenomena — from molecular geometries and spectroscopic signatures to macroscopic material properties. Covalent bonding, being highly directional, is responsible for substantial energy barriers to structural rearrangement. This results in pronounced metastability and the formation of rigid three-dimensional networks that exhibit exceptional mechanical properties. In contrast, the non-directional character of metallic bonding promotes high-coordination close-packed arrangements with low-barrier structural rearrangement, and yielding materials with characteristically low shear resistance and hardness.
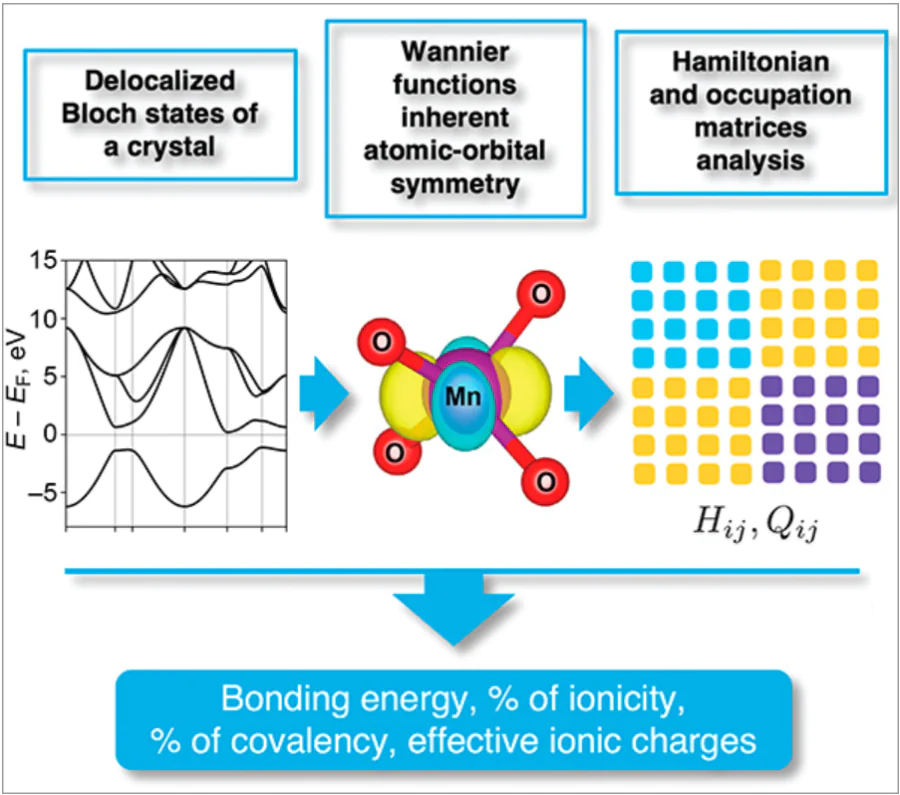
Quantitative characterization of chemical bonding — its ionic vs. covalent nature — represents one of the most persistent challenges in theoretical chemistry. Numerous approaches have been developed, including Löwdin and Mulliken population analyses, Bader’s Atoms in Molecules method, Natural Population Analysis, and Crystal Orbital Hamilton Population (COHP) analysis. Each method offers particular advantages, but suffers from limitations such as basis‑set dependence (for population analysis), sensitivity to the choice of integration domains or partitioning schemes, or reduced interpretability for crystalline systems. Here we present a review of a theory based on the Wannier function (WF) formalism, which enables first-principles quantum-mechanical calculation of both ionic and covalent contributions to the cohesive energy, as well as atomic charges.[1-4] The following sections illustrate these concepts with representative examples, while also highlighting the limitations and nuances of such classifications in real-world systems.
2. Foundations of the method: beyond formal oxidation states
Consider a binary compound AB. Its binding energy, Ebond, is typically defined as the difference between the total energy of the compound per formula unit (f.u.) at its equilibrium volume V0 and the sum of the energies of the free atoms A and B at infinity (Eq. 1):
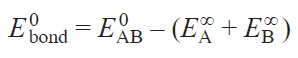
Solving the density functional theory (DFT) problem, we obtain a set of electronic eigenvalues and eigenfunctions,
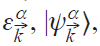
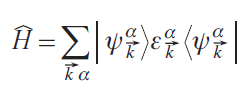
The corresponding density matrix operator is
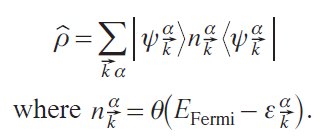
Wannier functions |Wi⟩ are calculated through a unitary transformation of the set of Bloch functions
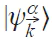
where |ji⟩ are trial atomic wave functions, and thus, |Wi⟩ also possess the same atomic orbital symmetry.
Then a basis set |Wi⟩ defined by Eq. 4 is orthonormalized. One can say that the Wannier functions |Wi⟩ are ’natural’ atomic orbitals for the electrons in a crystal, and the index i runs over the atomic quantum numbers nl (1s, 2s, 2p, 3p, 3d, ...). Equation 4 is one of the possible choices for the unitary transformation of the Bloch function set |k⟩, chosen to obtain WFs having the symmetry of the atomic orbitals, as is usually done in the analysis of chemical bonding. It is known that WFs are not uniquely defined. The projection procedure in Eq. 4 solves this problem, giving uniquely defined WFs that are most similar to atomic orbitals, are fully compatible with both the atomic limit and solid-state calculations, and allow us to separate the intraatomic and bonding effects.
Wannier functions |Wi⟩ from Eq. 4 are in real space representation. Sometimes, it is useful to define the reciprocal space representation
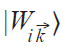
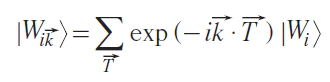
where T→ is the translation vector, i is an atomic number in the crystal unit cell, and a is a band number. One can define the Hamiltonian and density matrix in the WFs basis (Eqs 6, 7):

Then, the electronic energy E is:
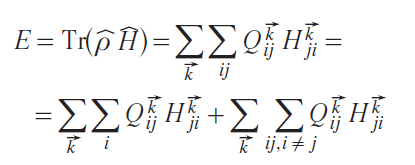
Separating the electronic energy E in Eq. 8 into covalent and ionic parts is not a trivial task. Although the interatomic term
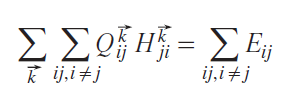
is clearly a covalent energy, the diagonal term
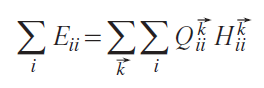
contains both contributions: the covalent energy for all atoms of type i in the crystal and the ionic part of the energy. To separate them, let us introduce the average energy
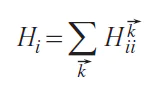
and the average occupancy
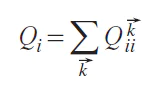
for atom i. The ionic part can be defined as Eiion = QiHi (Eq. 9), and the covalent part can be defined as Eicov = Eii – QiHi (Eq. 10). The electronic energy E in Eq. 8 can be written as Eq. 11:
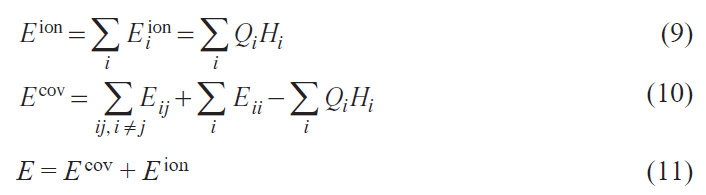
In a general case with orbital indices L = (l, m), Eq. 8 is
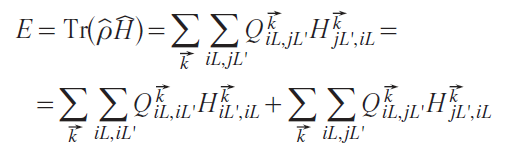
For the binary compound AB, the binding energy (see Eq. (1)) is
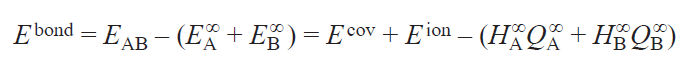
The following approximations could be useful:

Then, from Eqs 12 and 13,
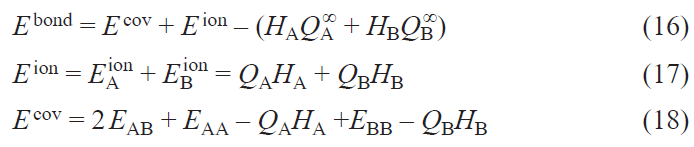
Hence, the binding energy separation is
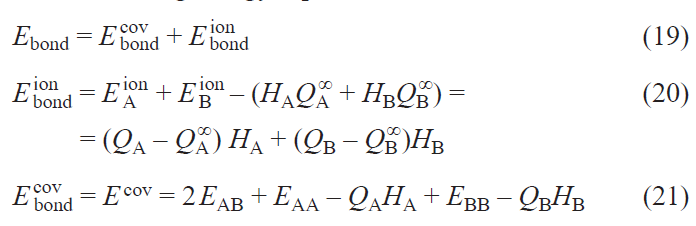
The covalent contribution to the binding energy E covbond in Eqs 18 and 21 contains, by definition (see Eq. (12)), only off-diagonal terms of the Hamiltonian and density matrices in the basis of WFs with atomic orbital symmetry (see Eq. 7). Hence, it corresponds directly to the common chemical understanding of a covalent bond between atomic orbitals. However, the ionic part is defined by the contribution of the diagonal terms of these matrices minus the HAQA∞ + HBQB∞ term. Hence, it contains not only the ion-ion interactions in the crystal, but also the energy of intraatomic electronic redistribution between different orbitals and also the energy of formation of charged ions from neutral atoms.
It is important to note that the energy E = Tr(ρ̂Ĥ) in Eq. (12) is not the total DFT energy but a sum over the occupied one-electron eigenvalues
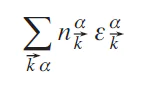
and so, unlike the total energy, it cannot be rigorously used to calculate energy differences (e.g., when computing energies of chemical reactions). Unfortunately, there is no way to split the total energy into such contributions as the ionic and covalent parts as it is done in Eqs 19 – 21. So, in the following we use the definition of Eqs 19 – 21 for the ionic and covalent parts of the one-electron energy Eq. (12). It is also useful to calculate the covalent bond energy separately for a given pair of atoms ij as
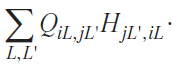
3. Quantitative analysis of chemical bonding
3.1. Beyond formal charges: covalent nature of MnO4– ion
Transition metals in high oxidation states often exhibit anomalous behavior, including metal-insulator transitions, charge ordering, and unconventional antiferromagnetism. Examples include Ni3+ in RNiO3 (see Ref. [5]) and Fe4+ in CaFeO3 (see Ref. [6]), where charge disproportionation can yield even higher formal valences (Ni4+, Fe5+), driven by strong metal — oxygen hybridization. These compounds are highly reactive due to their chemical instability, with the energy of formation of highly charged ions (e.g., 119 eV for Mn7+ in KMnO4) vastly exceeding typical bond energies (~ 10 eV), making their existence puzzling at first.
KMnO4 crystallizes in the orthorhombic Pmmn space group with unit cell parameters a = 5.93 Å, b = 7.58 Å, c = 9.23 Å. The Mn ion in KMnO4 is surrounded by four oxygen ions with a tetrahedral environment (corresponding to the Td point group symmetry) (Fig. 1). For this symmetry, five d-orbitals transform according to a triply degenerate irreducible representation t2g (orbitals xy, xz, yz) and doubly degenerate representation eg (orbitals 3z2 – r2 and x2 – y2). Unlike in octahedral symmetry, the t2g level lies higher in energy than the eg level.
![[{"id":"_SYIHK2YGY","type":"paragraph","data":{"text":"Crystal structure of KMnO<sub>4</sub>, consisting of MnO<sub>4</sub> tetrahedrons and space-filling potassium ions. Reproduced from Anisimov et al.<sup>2</sup> under the Creative Commons Attribution 4.0 International License."}}]](/storage/images/resized/m9crDCMjekieat7qUrl40CpkeRI8V36wFtWVZs1K_xl.webp)
The calculated band structure (Fig. 2) and density of states (DOS) (Fig. 3) show narrow electronic bands, reflecting the isolated nature of MnO4– units in the lattice. The partial DOS reveals two anti-bonding Mn d and O p hybridized states at 1 eV (eg) and 3eV (t2g), with their bonding counterparts at –5 eV. Between –4 and –1 eV, oxygen-derived non-bonding p-states dominate, showing negligible p – d hybridization.
![[{"id":"QrxUjjr9DW","type":"paragraph","data":{"text":"Band structure of KMnO<sub>4</sub>. Reproduced from Anisimov et al.<sup>2</sup> under the Creative Commons Attribution 4.0 International License."}}]](/storage/images/resized/Au2Do90YUMcXi1YuUFOYR241z9Eq9E6Nlc7RPTUY_xl.webp)
![[{"id":"laDB0wZA1w","type":"paragraph","data":{"text":"Density of states of KMnO<sub>4</sub>. Reproduced from Anisimov et al.<sup>2</sup> under the Creative Commons Attribution 4.0 International License."}}]](/storage/images/resized/f53tqRjPL6n1enLExTWyE8BFPPkQmSFoIhC5DLmx_xl.webp)
Wannier function analysis of the unoccupied states (Fig. 4) reveals a significant hole localization (50%) on the oxygen ligands (Table 1). Despite the formal Mn7+ (d0) configuration, oxygen p-orbitals dominate the hole states (50%), with only 4.54 d-holes (d 5.46 occupancy). This agrees with DFT-derived occupancy, indicating an effective Mn charge close to +2, which is substantially reduced from the nominal +7 value.
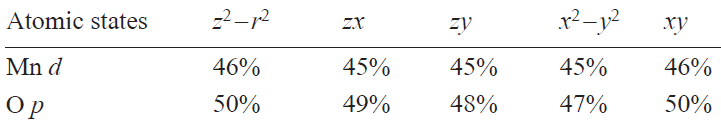
![[{"id":"9-DnaBYxOu","type":"paragraph","data":{"text":"Wannier functions (isosurface of the squared moduli) with the symmetry of manganese <i>d</i>-orbitals. Reproduced from Anisimov et al.<sup>2</sup> under the Creative Commons Attribution 4.0 International License."}}]](/storage/images/resized/sJJhD7ohrkbP8bzYI8Rwt6gJ3ytYH95GpKunJrwE_xl.webp)
Keeping in mind that there is ionic bonding between K+ and MnO4– ions in KMnO4, we calculated the contribution of the covalent and ionic parts to the chemical bond within the MnO4– complex according to Eq. 18. The obtained values E covbond = –38.2 eV and E ionbond = 0.07 eV confirm that MnO4– ion in KMnO4 is a totally covalent complex. In the following, we map the picture of chemical bonding in KMnO4 onto a simple model. Chemical bonding, traditionally described through localized atomic orbitals, requires generalization in crystalline systems, where electrons form delocalized Bloch states. Wannier functions bridge this gap, providing localized, symmetry-adapted orbitals via unitary transformations of Bloch waves.
For an AB compound, the bonding can be captured by a minimal Wannier-based Hamiltonian, parametrized by the difference of the orbital energies of atoms A and B (ΔE = HA – HB), governing charge transfer (ionic character) and hopping integrals (HAB = t), determining orbital mixing (covalent character).
This framework enables systematic analysis of bonding mechanisms, from purely ionic to strongly covalent limits, and provides a benchmark for first-principles calculations.
Consider atoms A and B, each contributing a single partially filled nondegenerate orbital (total occupancy: 2 electrons). The system Hamiltonian in the Wannier basis reduces to:
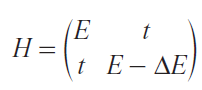
where E is the energy level of the first orbital (the higher one) and second ion has the orbital energy lower by ΔE. The hopping energy term t corresponds to hybridization between the orbitals.
From the eigenvectors of the model Hamiltonian (Eq. (22)) one can get occupation numbers for the two atoms (A and B) orbitals (Q1 and Q2) and then calculate the charge transfer Q2 – Q1 from the first to the second atom. One can easily see that the larger t/ ΔE, the smaller charge transfer Q2 – Q1 that characterizes ionicity and hence the chemical bond in the system will be more covalent. On the contrary, if the value of t/ΔE parameter is small, then the opposite is true and the system will be characterized by mostly ionic bonding.
Atomic charges can be calculated from the charge transfer Q2 – Q1 as follows. If Q1inf and Q2inf are orbital occupations for the corresponding neutral atoms A and B, then the charges of the atoms in the compound are: Z1= Q1inf – Q1, Z2= Q2inf – Q2. To keep electroneutrality of the system: Z2 = (Z2 – Z1)/2. Then Z2 = [(Q2inf – Q1inf)(Q2 – Q1)]/2. Since for our simple model Q1inf = Q2inf = 1, one gets that Z2 = (Q1 – Q2)/2.
To analyze chemical bonding in KMnO4, we constructed symmetry-adapted WFs for the MnO4– complex. For each Mn d-orbital, we formed linear combinations of O p-orbitals sharing the same irreducible representation (Td point group), yielding five maximally hybridized p – d orbital pairs. This generates a 10-dimensional bonding subspace, while the remaining seven orthogonal O-p combinations form non-bonding states.
The resulting orbital partitioning directly maps to the electronic structure: the non-bonding subspace (–4 to –1 eV) and bonding/antibonding states (–5 eV and 1 – 3 eV, respectively) are clearly resolved in the band structure and DOS (Fig. 2, Fig. 3). This approach indicates the covalent (p – d hybridized) and ionic (non-bonding) contributions within a unified framework.
Those 17 orbitals were used to build 17 WFs that describe all occupied and empty energy bands, i.e. all bands within the [–6; 4] eV energy interval corresponding to MnO4– ion.
The separation of the electronic states into bonding and non-bonding subspaces is clear (see Fig. 2, Fig. 3). We have calculated within this new basis the Hamiltonian matrix and the occupation matrix. These 10 × 10 matrices, due to the presence of the rather high symmetry of MnO4– tetrahedra, could be constructed by doubly degenerate 2 × 2 matrices that describe interaction of the Mn-eg states with the O-p bonding orbital and triply degenerate 2 × 2 matrices for the Mn-t2g states and the corresponding O-p bonding orbitals with off-diagonal elements representing overlap occupations:
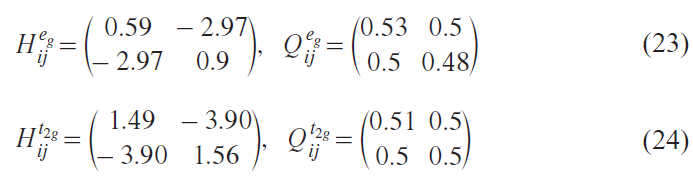
The 10 × 10 bonding space decomposes into five orthogonal 2 × 2 subspaces, each well described by Eq. (22). Diagonalization yields eigenvalues of 3.72/–2.23 eV (t2g) and 5.48/–2.43 eV (eg), matching the antibonding/bonding states in Fig. 3. The occupation matrices reveal nearly symmetric charge distributions Q2 – Q1 ≈ 0).
Through Wannier function analysis of KMnO4, we demonstrate that chemical bonding in the MnO4– complex is predominantly covalent with negligible ionic contribution, where the calculated Mn-3d electron occupancy of approximately five electrons indicates an effective Mn2+ like state rather than the formal Mn7+ configuration. This electronic structure is consistent with a d2L2 configuration, a notation denoting a correlated state of two localized d-electrons and two ligand-holes, which involves oxygen holes — localized unpaired electrons predominantly residing on oxygen 2p orbitals that create O– or O0-like character rather than formal O2– states.
3.2. Driving force for the formation of ternary oxides
The driving forces behind compound formation have been a central question in chemistry since the early 19th century, when Davy and Berzelius first proposed that ‘electropositive’ (metallic) and ‘electronegative’ (nonmetallic) elements are attached to each other to form stable compounds. This concept later evolved into the modern theory of electronegativity, where a large difference in electronegativities between elements stabilizes chemical bonding through electron transfer, which is consistent with Mulliken’s definition of electronegativity as minus the electron’s chemical potential.
When considering more complex systems, such as ternary and quaternary compounds, classical acid – base theory provides partial insights. Basic oxides like CaO and Na2O react with acidic SiO2 but not with each other. Bases are electron donors, while acids are acceptors. However, the exact mechanisms of electron redistribution remain unclear in cases where all bonding orbitals are occupied, as in closed-shell oxides.
In this Section, the proposed approach, after successful validation on binary compounds, is applied to more complex systems including ternary oxides.[2]
In addition, it focuses on three representative cases of ternary oxide formation, viz., the reaction of 2 MgO with SiO2 to form Mg2SiO4; MgO with Al2O3 forming MgAl2O4; and Na2O with SO3 yielding Na2SO4. The analysis concentrates on fundamental changes in bonding characteristics during these transformations, examining both the energies of formation and electronic structure evolution, while deliberately excluding consideration of thermodynamic conditions or kinetics.
3.2.1. Mg2SiO4
Mg2SiO4 crystallizes in the olivine structure (space group Pnma); its structure is shown in Fig. 5с. It is formed from MgO and SiO2 via 2 MgO + SiO2 → Mg2SiO4, maintaining the original coordination numbers: octahedral for Mg (6 neighbors, see Fig. 5a) and tetrahedral for Si (4 neighbors, see Fig. 5b).
![[{"id":"SgXIDIsD8t","type":"paragraph","data":{"text":"Crystal structures of MgO (<i>a</i>), SiO<sub>2</sub> (<i>b</i>), Mg<sub>2</sub>SiO<sub>4</sub> (<i>c</i>), Na<sub>2</sub>O (<i>d</i>), SO<sub>3</sub> (<i>e</i>), MgAl<sub>2</sub>O<sub>4</sub> (<i>f</i>), α-Al<sub>2</sub>O<sub>3</sub> (<i>g</i>), Na<sub>2</sub>SO<sub>4</sub> (<i>h</i>).<sup>3</sup> Red spheres correspond to O atoms."}}]](/storage/images/resized/fKHxS95is1R9yksGBp080jIMsWxiv8YttuxQDPbV_xl.webp)
In all three compounds, the top of the valence band is dominated by O-p states hybridized with Mg/Si-s/p, while conduction band bottoms contain mainly Mg/Si-s/p states. These states form our WF basis.
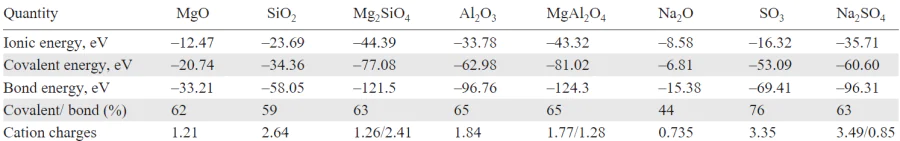
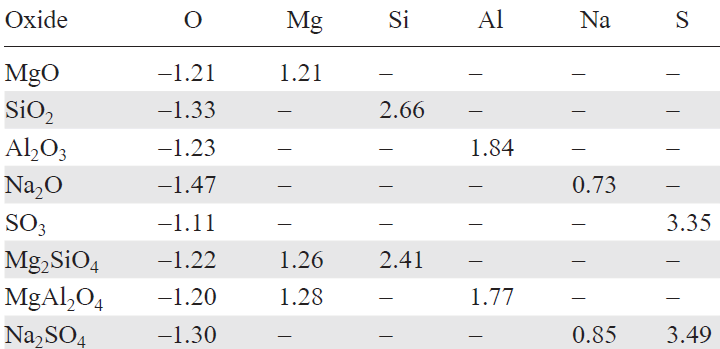
DFT calculations show that the formation of Mg2SiO4 from oxides is exothermic by 0.575 eV/f.u. (0.082 eV/atom), despite opposite one-electron energy trends. The covalency increases slightly: 62% (MgO) → 59% (SiO2) → 63% (Mg2SiO4).
We analyzed atomic charges (Table 2, Table 3) to track electron redistribution during the formation of the complex compounds. In Mg2SiO4:
— Mg charge increases from ZMg = 1.21 (MgO) to 1.26 (0.05 e– transfer per Mg, 0.10/f.u.),
— Si charge decreases from ZSi = 2.64 (SiO2) to 2.41 (0.23 e– gain),
— O charges change from ZO = –1.32 (SiO2) and –1.21 (MgO) to –1.22/–1.28.
The net charge analysis shows 0.055 e– transfer from 2 MgO to SiO2, consistent with the acid-base theory. Bond energies are strengthened in the ternary oxide:
— EMg – O: –2.93 eV (MgO) → –3.096 eV (Mg2SiO4),
— ESi – O: –8.267 eV (SiO2) → –9.359 eV (Mg2SiO4).
3.2.2. MgAl2O4
MgAl2O4 crystallizes in the spinel structure (space group Fd3̅m; see Fig. 5f ). Key structural differences from precursor oxides are as follows:
— Al maintains octahedral coordination (both in Al2O3 and spinel),
— Mg changes from octahedral (MgO) to tetrahedral coordination.
— Bond lengths change significantly:
— Mg – O: 2.095 Å (MgO) → 1.938 Å,
— Al – O: average 1.913 Å (1.855/1.972 Å) → 1.944 Å.
The formation of MgAl2O4 is energetically favorable by 0.2328 eV/f.u. (0.033 eV/atom), though the small magnitude suggests weak driving forces, with one-electron energies again showing opposite trends. The covalency remains nearly constant: 65% (Al2O3), 62% (MgO), and 65% (MgAl2O4).
Charge analysis (see Tables 2, 3) reveals electron redistribution: Al gains 0.07 e– (ZAl = 1.84 → 1.77), while Mg loses 0.07e– (ZMg = 1.21 → 1.28). Oxygen charge decreases in magnetude slightly (ZO = –1.235 in Al2O3, –1.21 in MgO, –1.20 in spinel), consistent with the ~1 – 2 eV energy difference between Al and Mg states.
Bonding analysis shows strengthening interactions:
— Al – O: average ZAl – O = –4.948 eV (Al2O3, with –5.514/–4.383 eV for short/long bonds) → –5.06 eV (spinel, equal-length bonds),
— Mg – O: ZMg – O = –2.93 eV (MgO) → –3.35 eV (spinel), despite reduced coordination (6 → 4 bonds/Mg).
The net 0.07 e– transfer from MgO to Al2O3 agrees precisely with the acid-base theory predictions.
3.2.3. Na2SO4
Na2SO4 (see Fig. 5h) exhibits predominantly ionic bonding between Na+ and (SO4)2– ions, with strong covalent S – O bonding within the sulfate group. The formation of Na2SO4 from Na2O and SO3 is highly exothermic (4.914 eV/f.u., 0.702 eV/atom), with one-electron energies correctly predicting the trend, albeit overestimating by more than two times. WFs were constructed using O-p, S-s/p, and Na-s states.
Charge analysis (see Tables 2, 3) reveals changes related to the formation of Na2SO4 from Na2O + SO3:
— Unexpectedly, sulfur charge increases (ZS = 3.35 → 3.49), with oxygen charge –1.30 in Na2SO4 on average vs. –1.47 in Na2O and –1.11 in SO3,
— Each Na atom loses 0.115e– (ZNa = 0.735 → 0.85),
— Each SO3 unit gains 0.23e–, and each Na2O loses 0.23 e– consistent with acid-base theory.
The WF formalism reveals that the formation of complex compounds from simple oxides follows Lewis acid-base principles at the quantum mechanical level, with electron density transferring from high-energy orbitals of basic components (e.g., Na2O) to lower-energy orbitals of acidic partners (e.g., SO3). This redistribution occurs through modulation of bond ionicity rather than oxidation state changes. Reactivity trends are systematically correlated with electronegativity differences, as evidenced by the series of increasing average electronegativity Na2O → MgO → Al2O3 → SiO2 → SO3, where the most exothermic reaction occurs between extreme members (Na2O + SO3) and the least pronounced between adjacent pairs (MgO + Al2O3).
Our analysis quantitatively establishes that such acid-base charge redistributions consistently enhance overall bonding in the resulting ternary oxides, providing a unified explanation for their thermodynamic stability. The methodology offers a powerful bridge between classical chemical concepts and first-principles quantum mechanical descriptions of electron behavior in condensed matter systems.
3.3. Does covalency decrease with coordination number?
The subsequent analysis of ZnO, CO2, and NaCl structures, based on our earlier study employing the pairwise covalency decomposition approach,[1] challenges the traditional assumption that covalency decreases and ionicity increases with coordination number. This conclusion follows from both Bader analysis and Wannier function-based charge definitions.
ZnO exists in two main phases: wurtzite (Wrzt) with tetrahedral fourfold coordination and rocksalt (RS) with octahedral sixfold coordination (see Fig. 6a,b). The equilibrium Wrzt phase transitions to the cubic phase under moderate pressure (~9 GPa), commonly attributed to reduced lattice dimensions enhancing Coulomb interactions and favoring ionic bonding. This transition, traditionally thought to reflect a shift from covalent to ionic character, has been extensively studied using first-principles calculations.[7-11]
![[{"id":"hfTnmx0UPC","type":"paragraph","data":{"text":"Crystal structure of ZnO in wurtzite (<i>a</i>) and rocksalt phase (<i>b</i>), CO<sub>2</sub> in molecular cubic (<i>c</i>) and β-cristobalite (<i>d</i>) phases. Brown, green and blue spheres are Zn, C and oxygen atoms, respectively.<sup>4</sup>"}}]](/storage/images/resized/ETXQF62sNIRSfvvVrEbt7NrMEu5cnvhs8fzcp7tF_xl.webp)
The degree of ionicity and covalency within the WF formalism is not directly measurable experimentally and depends on the choice of the basis set. To obtain an independent reference, we also performed a Bader analysis, which partitions the total charge density into atomic contributions by integrating over regions bounded by zero-flux surfaces of the charge density gradient. This approach provides a first-principles estimate of ‘natural’ ionic charges. Within these regions, the charge density decreases from the atomic center to the boundary before increasing again, reflecting the redistribution of the electronic density between atoms.
The Wannier function analysis yields consistent results: ZZn = +0.92 and ZO = –0.92 for the cubic phase, and ZZn = +0.97 and ZO = –0.97 for the Wrzt phase. Notably, the direction of charge variation contradicts the conventional expectation: ionicity decreases in the cubic phase relative to the tetrahedral phase.
In the Wannier function formalism, ionic charges are determined by the occupancy of the corresponding Wannier function Wi, which is obtained by projecting the atomic orbital φi onto the Bloch function (see Eq. 4). The function Wi contains a fraction xi of the charge of the atomic orbital φi, while the remaining charge is distributed among neighboring atoms. Therefore, to obtain the charge associated with φi, the occupancy of Wi must be multiplied by the factor xi.
Applying this renormalization yields ZZn = +1.11 for ZnO in the cubic phase and ZZn = +1.10 in the Wrzt phase. These values show excellent agreement with the results of the Bader analysis.
The results of the calculations of the WFs are presented in Table 4.
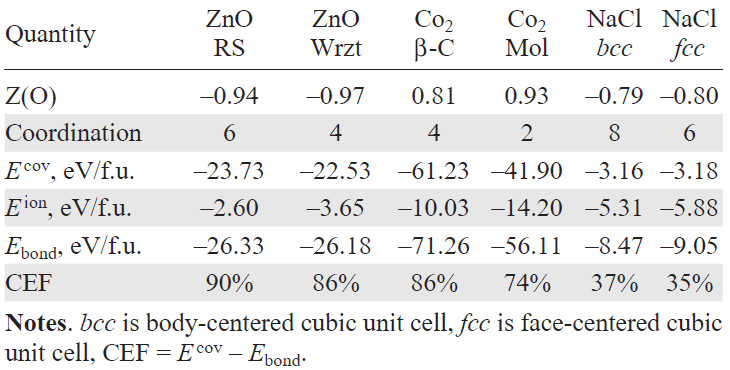
Here, Epair-bond represents the covalent energy of a single metal–oxygen bond. In the tetrahedral Wrzt phase, where the metal-oxygen bond length is shorter than in the cubic phase, individual bonds exhibit a higher energy: Epair-bond = –3.2 eV in the cubic phase and Epair-bond = –5.2 eV in the Wrzt phase.
Although the Wrzt phase has only four nearest neighbor bonds compared to six in the cubic phase, the total covalent energy contribution from these nearest neighbors is greater in the Wrzt phase. However, the overall bonding energy, which includes contributions from more distant coordination spheres, is slightly lower in the Wrzt structure due to its lower packing density. The extended nature of Wannier functions, which enhances hybridization with atoms beyond the first coordination shell, may contribute to a slight overestimation of the covalency in the cubic phase. However, the total covalent energy in both phases remains comparable, with a minor energy gain in the cubic phase attributed to a slight increase in ionic character. This contradicts the conventional expectation that the ionicity should increase in the cubic phase.
The minimal change in ionic charges during the phase transition is further confirmed by Bader analysis. For the RS phase, the calculated charges are ZZn = +1.20 and ZO = –1.20, while for the Wrzt phase they are ZZn = +1.19 and ZO = –1.19. Thus, the variation in ionicity across the phase transition is negligible and within the margin of computational accuracy. The WF approach yields consistent results: ZZn = +0.92 and ZO = –0.92 for the RS phase, and ZZn = +0.97 and ZO = –0.97 for the Wrzt phase.
CO2 is known for its linear molecular structure in the gas phase. Under pressure, it transforms from the cubic Pa3̅ (‘dry ice’) phase to the Cmna phase at 10 GPa, followed by a pseudotetragonal (P42/mnm or Pnnm) phase at 20 GPa, and finally, above 35 GPa and 1800 K, transforming into the polymeric β-cristobalite phase[12][13] (see Fig. 6d ).
Similarly to ZnO, CO2 experiences an increase in coordination number under pressure. In the low-pressure (LP) cubic Pa3– phase, carbon has two nearest oxygen neighbors (twofold coordination of C and O). In the high-pressure β-cristobalite phase, the carbon atom is positioned at the center of an oxygen-coordinated tetrahedron (fourfold coordination of C and two-fold coordination of O).
Bader charge analysis shows a slight reduction in ionicity and a corresponding increase in covalency upon the transition from the molecular cubic (ZC = +2.14, ZO = –1.06) to the β-cristobalite phase (ZC = +1.96, ZO = –0.98), contrary to the expected trend with increasing coordination. Wannier function results confirm this tendency (molecular cubic: ZC = +1.85, ZO = –0.93; β-cristobalite: ZC = +1.63, ZO = –0.81), as summarized in Table 4.
Here, Epair-bond denotes the covalent energy of one C – O bond. In the β-cristobalite phase (fourfold coordination), the C – O bond length increases to 1.402 Å compared to 1.171 Å in the cubic Pa3̅ phase (twofold coordination). Nevertheless, the bond energy per pair is less negative in the β-cristobalite phase (–14.46 eV) than in the molecular cubic phase (–17.73 eV), while the higher coordination leads to a larger total covalent contribution. The ionic term decreases, as reflected by reduced atomic charges in both Wannier and Bader analyses, similar to ZnO.
The WF formalism also allows estimation of covalent energies beyond the first coordination shell. In molecular CO2, the O – O covalent energy within one molecule is about –1.60 eV, whereas between molecules it ranges from –0.42 eV to –0.74 eV, depending on orientation. In the third shell, the O – O energy is nearly constant at ≈ –0.68 eV, indicating that at such distances it is governed mainly by WF overlap and symmetry. Though weaker, these long-range terms still contribute to the cohesive energy.
The calculated bonding energies differ from atomization energies, as they omit repulsive overlap effects, which are included in the total DFT energy Etot. For CO2, their magnitude exceeds that for ZnO and correlates with interatomic distances.
NaCl, a textbook ionic compound, crystallizes in the ambient-pressure RS structure with sixfold Na – Cl coordination. Under high pressure, it adopts the CsCl-type phase with eightfold coordination.[14][15] The corresponding WF results are also summarized in Table 4.
Here, Epair-bond is the covalent energy of a single Na – Cl bond. Unlike in CO2, it depends on bond length: in the NaCl-type phase (dNa – Cl = 2.851 Å), Epair-bond = –0.50 eV, while in the CsCl-type phase (dNa – Cl = 3.038 Å) it decreases to –0.38 eV. As in ZnO and CO2, covalency shows a slight increase with coordination number.
The common belief that lower coordination numbers correspond to greater covalency and reduced ionicity rests on several oversimplified assumptions. First, covalent bonding is often described as a set of two-center, two-electron bonds, implying that coordination numbers above four are unfavorable, but this idea is contradicted by examples such as boranes, where multicenter bonding is essential. Second, while covalent interactions are typically directional and ionic ones non-directional, coordination numbers in ionic compounds are mainly dictated by ionic radii ratios: low coordination can occur in ionic systems with significant size mismatch, and high coordination is possible in covalent systems with multicenter bonds. Third, the short-ranged nature of covalent bonding and the long-ranged Coulomb nature of ionic bonding commonly thought to lead to more ionic bonding in close-packed structures, but there is no reason why they will do not preclude strong covalency in densely packed structures.
From an electronegativity perspective, electrons transfer from less to more electronegative atoms until their electronegativities equalize.[16] Since both electronegativity and its dependence on charge are intrinsic atomic properties, the resulting charges should be largely independent of crystal structure. What matters is the nature of constituent atoms and their proportion.
3.4. Ionicity vs. charge
Table 5 and Table 6 present the results obtained using the WF approach to evaluate ionicity, based on the atomic charge distributions and the decomposition of the cohesive energy into ionic and covalent contributions for a series of simple compounds.
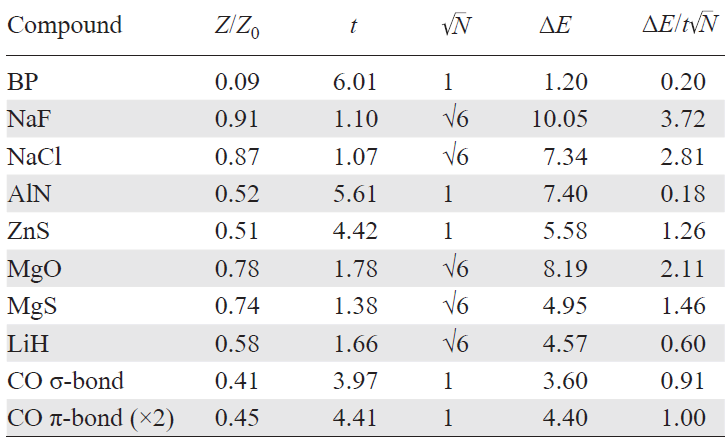
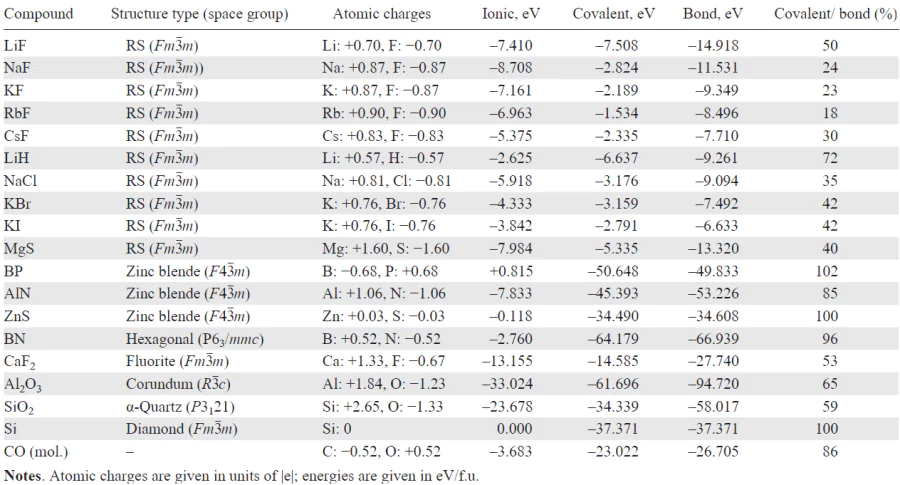
The analysis reveals several key trends: 1) both the degree of ionicity (defined as a ratio of effective to formal valence charges) and the ionic-to-covalent energy ratio increase with growing electronegativity difference. 2) Boron phosphide BP in the zinc blende structure shows anomalous behavior with inverted atomic charges (B: –0.68, P: +0.68), leading to a covalency degree above 100% and a destabilizing (positive) ionic contribution — indicative of unfavorable charge transfer that nonetheless enhances covalency. 3) Similar charge inversion is seen in the CO molecule, consistent with the Zintl – Klemm rule: oxygen donates an electron to carbon, enabling a C–≡O+ configuration with stronger covalent bonding (without such charge transfer only a double bond would be possible, C=O). 4) Across all compounds, effective charges are significantly smaller than formal ionic valences.
All results presented in Table 5 for compounds with various bonding types can be qualitatively understood within a simple two-orbital model as shown in Eq. (22). The essential parameters in this model are the difference of on-site (atomic) energies ΔE = HA – HB, where HA and HB are the diagonal elements of the Hamiltonian and the off-diagonal matrix element t = HAB, which represents the hybridization (hopping) between orbitals (see Eq. (22)).
The result for BP, showing reversed atomic charges (+0.68 on phosphorus and –0.68 on boron), is counterintuitive and demands explanation, as phosphorus is more electronegative than boron on all standard scales. This apparent contradiction can be understood as follows. In a purely covalent picture, neutral B and P atoms each form three 2-center 2-electron bonds. However, if one considers inverse charges (B– and P+) according to the Zintl – Klemm rule, each atom can form four such covalent bonds, thereby strengthening the bonding network. If the gain in covalent stabilization outweighs the energetic cost of this charge inversion (reflected in a positive ionic contribution to the cohesive energy, see Table 5 and Table 6), such an unusual charge distribution becomes energetically favorable.
Furthermore, an analysis of WFs shows that the bonding molecular orbital remains asymmetric and is shifted toward the P atom, in agreement with its higher electronegativity (Fig. 7). Orbital occupation numbers support this view: boron has 3.68 valence electrons, phosphorus has 4.32. However, relative to their neutral states (3 and 5 valence electrons, respectively), this corresponds to formal charges of –0.68 (B) and +0.68 (P). These are static charges arising from bonding-induced charge redistribution. Notably, similar charge inversions are observed in Born effective charges of BP and related compounds.[17-19]
![[{"id":"H_dpHIXASF","type":"paragraph","data":{"text":"Section of the squared Wannier functions centered on the boron (<i>a</i>) and phosphorous (<i>b</i>) ions. Section of the squared bonding molecular orbital (<i>c</i>)."}}]](/storage/images/resized/2oU0XiBg2VqcG0GnYlxHj2tCKppLttNGYt7ZlE9d_xl.webp)
This example demonstrates that atomic charges in covalently bonded systems can diverge significantly from expectations based solely on electronegativity. In BP, the competition between ionic energy and covalent bond enhancement leads to the observed inverted charge distribution.
For the two-orbital model we can define the degree of ionicity as the ratio of the effective atomic charge to the formal charge: Z/Z0.
As shown in Fig. 8, this ratio raises with increasing ratio ΔE/t, indicating that stronger hybridization (larger t) leads to a more covalent character of the bond. Thus, the degree of ionicity is inversely related to the degree of hybridization between atomic orbitals, whereas increasing ΔE (difference of electronegativities) increases ionicity (see Eq. (22), Table 5).
![[{"id":"bC7QCTRVim","type":"paragraph","data":{"text":"Ratio of the effective atomic charge to the formal charge (|Z/Z<sub>0</sub>|) for the model. Mapping of the results of DFT calculation onto the two-band model (symbols)."}}]](/storage/images/resized/MH1USwXUEwIvLoWneaKHdyGkGMhr8xyFdOLliuEr_xl.webp)
We first apply the two-orbital model to LiH, which closely matches its assumptions: both Li and H possess a single s-orbital occupied by one electron in the neutral atom. The main distinction is structural — each atom in the RS lattice forms six equivalent bonds with its neighbors, so the number of hybridization channels is N = 6. Within perturbation theory, this multi-channel hybridization can be approximated in the two-orbital model by an effective hopping term Nt (see Table 5).
The model parameters E, ΔE, and t are extracted from the diagonal and off-diagonal elements of the Wannier-based Hamiltonian for LiH. The resulting Z/Z0 agrees well with the results of our calculations.
A similar analysis was carried out for other RS compounds NaF, NaCl, MgO, and MgS yielding similarly good agreement. These systems are predominantly ionic, with ΔE/t > 1.
For semiconductors with the zinc blende structure, such as AlN, BP, and ZnS, we evaluated formal charge for a single sp3 orbital pair directed along the bond axis, to map it to the two-orbital model. For the BP case, |Z/Z0| is utilized since the simple two-orbital model does not support inverted charges. Here, the hopping term t corresponds to the strongest orbital overlap. These materials also follow the model curve closely, confirming their mixed ionic–covalent bonding character.
3.5. Charge transfer vs. energetics
Fig. 9 summarizes the data with quantitative characterization (R = E cov/Ebond) of the chemical bond type for a series of compounds considered. Clear correlation shows that the proposed method based on WFs gives a consistent correlation linking charge transfer and the energies of chemical bonds. The ionic energy must be proportional to the square of the degree of ionicity, so the percentage of the ionic contribution to the bond energy should be defined by the value of (Z/Z0)2, where Z0 is the formal valence. As shown in Fig. 9, this naive estimation is remarkably well satisfied in most cases.
![[{"id":"pPyZomGIg1","type":"paragraph","data":{"text":"Degree of ionicity (1 – E<sup>cov</sup>/E<sub>bond</sub>) as a function of squared number of electrons transferred between adjacent atoms normalized by formal valence."}}]](/storage/images/resized/Cuo1D7txlzVDuVXgJSki5HjhkzGwyIUxy2xlTFLe_xl.webp)
4. Conclusion
A comprehensive review of a first-principles approach for determining effective atomic charges and decomposing cohesive energy into ionic and covalent contributions using Wannier functions is presented. The method was systematically applied to a diverse set of materials covering ionic, covalent, and mixed-bonding regimes. A simplified model incorporating orbital energy differences and hybridization strength further validates our Wannier-derived charges. The approach proves robust across conventional and exotic systems, offering deep insights into chemical bonding mechanisms while maintaining quantitative accuracy. The presented Wannier‑function approach is not limited to crystalline insulators and semiconductors. It is applicable to defects, interfaces, and chemical reactions, providing a quantitative separation of ionic and covalent contributions to bond energies and reaction barriers. The ability of the approach to bridge qualitative chemical concepts with rigorous quantum‑mechanical calculations makes it a promising tool for a wide range of problems in chemistry and materials science.
This work was supported by the Russian Science Foundation (Grant No. 19-72-30043). D.Y.N., D.M.K., A.O.S., M.A.M., and V.I.A. also thank the Ministry of Science and Higher Education of the Russian Federation (state assignment of the IMP UB RAS).
The authors have no conflicts to disclose.
Author contributions
Dmitry Y. Novoselov: Writing the main manuscript text, review & editing; Investigation and analysis (equal). Dmitry M. Korotin: Formal analysis (equal); Investigation (equal); Software (lead); Writing — review & editing (equal). Alexey O. Shorikov: Formal analysis (equal); Investigation (equal); Writing — review & editing (equal). Mary A. Mazannikova: Formal analysis (equal); Investigation (equal); Writing — review & editing (equal). Vladimir I. Anisimov: Conceptualization (equal); Methodology (lead); Writing — review & editing (equal). Artem R. Oganov: Conceptualization (equal); Supervision (lead); Funding acquisition (lead); Investigation (equal); Project administration (lead); Writing — review & editing (equal).
Data availability statement
The data that support the findings of this study are available in the article.
5. List of abbreviations
bcc — body-centered cubic (unit cell);
CEF — covalent energy fraction;
COHP — Crystal Orbital Hamilton Population;
DFT — density functional theory;
DOS — density of states;
fcc — face-centered cubic (unit cell);
f.u. — formula unit;
RS — rocksalt (phase);
WF — Wannier function;
Wrzt — wurtzite (phase).
References





