



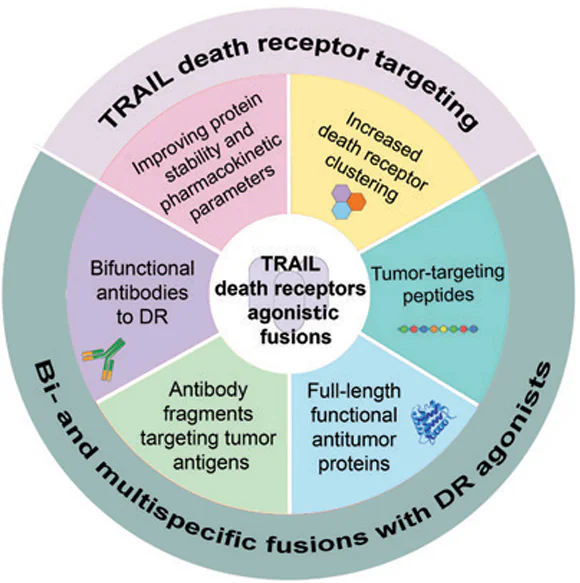
Keywords
Abstract
The development of therapeutic bispecific antibodies and hybrid proteins is one of the most urgent biomedical technologies with obvious clinical prospects. At the same time, advanced strategies of molecular design of drugs with new properties are coming to the forefront. The tumour necrosis factor-related apoptosis inducing ligand (TRAIL) receptor pathways are important components of the immune system involved in the immune surveillance and selective elimination of transformed cells. TRAIL-based proteins are therefore promising drug candidates for the treatment of malignant tumours and autoimmune diseases. In the first series of clinical trials, drugs targeting the death receptors DR4 or DR5, did not show significant anti-cancer activity. This is due to the TRAIL resistance mechanisms that tumours evolve to evade the efficient induction of apoptotic signalling. However, a wide range of novel TRAIL death receptor-targeted formulations are currently being developed, mainly to improve stability, enhance death receptor clustering and involve additional tumour targets. Over the past decade, several dozens of multi-targeted fusion proteins with either TRAIL protein or DR5-specific agonistic monoclonal antibodies have been developed to improve therapeutic efficacy. These include fusions with either short peptide tags or large functional proteins, as well as antibody fragments targeting molecular pathways involved in angiogenesis or proliferative signalling such as epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), programmed death-ligand 1 (PD-L1), etc. Collectively, these multimodal proteins enhance the activation of extrinsic and intrinsic apoptotic pathways in transformed cells, as well as affect the tumour microenvironment. This comprehensive review systematizes the bispecific and multivalent fusion proteins and conjugates targeting TRAIL death receptors, analyze the molecular mechanisms by which they overcome tumour resistance to TRAIL, and assess their clinical prospects.
The bibliography includes 236 references.
1. Introduction
Due to their ability to simultaneously activate several signalling pathways that affect tumour development, bispecific fusion proteins are on the cutting-edge of the modern anticancer drug discovery.[1] Among them, cytokines engineered for the selective cell targeting, including so-called ‘supercytokines’, ‘immunocytokines’, ‘fusokines’ and other synthetic cytokines, serve as an important platform for the development of novel protein-based therapeutics with enhanced biological properties.[2]
The cytokine TRAIL, a member of the tumour necrosis factor (TNF) family, is an important component of the immune system that selectively eliminates transformed and aberrant cells by apoptosis after binding to the death receptors (DRs) DR4 or DR5.[3][4] Unlike TNF and FAS ligands, TRAIL is the only natural cytokine that induces apoptosis in transformed cells without significant toxicity to normal tissues.[3][5] This has served as the basis for the development of DR agonists for the targeted therapy of tumour diseases. However, the first-generation DR agonist, soluble recombinant TRAIL alone or in combination with chemotherapeutic drugs, appeared to be ineffective in the clinic.[5-11] Similarly, the first monoclonal antibodies targeting DRs have also shown very limited anti-cancer effect.[12]
Like some other members of the TNF family, TRAIL can induce the production of pro-inflammatory chemokines and cytokines that promote a tumour-supportive immune microenvironment, which may counteract its antitumour activity.[13] There is also increasing evidence that the TRAIL DR signalling pathway is involved in the regulation of cancer invasion and metastasis, with both positive and negative roles being reported.[14-16] This is achieved by a complex balance between TRAIL-induced activation of pro-apoptotic signal transduction and non-canonical pro-survival pathways.[17][18] However, despite the failure of the first-generation DR agonists, the implementation of rational and sophisticated molecular design strategies has led to a new wave of promising developments, comprising of several dozen new molecular constructs targeting TRAIL DRs with enhanced agonistic activity, improved pharmacokinetics and therapeutic efficacy.[17] [19-24] This review aims to provide a comprehensive overview of the developed bispecific and multivalent fusion proteins and conjugates targeting TRAIL DRs with an assessment of their clinical prospects.
2. TRAIL signalling pathways
The cytokine TRAIL was identified by two independent groups in 1995 – 1996 on the basis of its sequence homology to TNF and FasL.[25][26] Analysis of the crystal structure of the TRAIL protein revealed that soluble TRAIL forms a homotrimer similar to other members of the TNF family.[27] The biological activity of TRAIL is critically dependent on the presence of an unpaired cysteine (Cys230), which is involved either in the formation of an interchain disulfide bridge, resulting in the formation of a poorly active TRAIL dimer, or in the formation of an active trimer by chelation of a single zinc atom.[28][29] TRAIL is expressed as a type II transmembrane protein, and its extracellular domain can be proteolytically cleaved from the cell surface presumably by a cysteine proteases to form a soluble ligand.[30][31]As soluble TRAIL has shown high biological activity, the bulk of the studies have been carried out using recombinant preparations of the extracellular domain of the protein containing 95 – 281 or 114 – 281 amino acids.
To date, four membrane-bound receptors have been identified, TRAIL-R1 (DR4), TRAIL-R2 (DR5), TRAIL-R3 (DcR1), and TRAIL-R4 (DcR2), which bind TRAIL with similar affinities and subnanomolar dissociation constants.[29] [32][33] The agonistic death receptors (DRs) DR4 and DR5 are type I transmembrane proteins and contain an intracellular death domain (DD) that normally stimulates apoptosis upon TRAIL binding.[34][35] DcR1 and DcR2 are transmembrane proteins that do not have a fully developed intracellular DD and are unable to transmit an apoptotic signal, so they are called decoy receptors.[36-39] In addition, decoy receptors compete with the apoptosis-inducing DR4 and DR5 for ligand binding and inhibit TRAIL-induced apoptosis.[40] TRAIL also showed high affinity for the secreted soluble decoy receptor osteoprotegerin (OPG), which lacks both transmembrane and cytoplasmic residues and attenuates TRAIL-induced apoptosis.[41] The guide tree and schematic structure of human TRAIL receptors are shown in Fig. 1.
![[{"id":"2DJMFbdfrm","type":"paragraph","data":{"text":" The guide tree and schematic structure of human TRAIL receptors. Sequences were aligned using Uniprot with the Clustal Omega program following Guide tree option."}}]](/storage/images/resized/bB8AnbJpSNARrr3ujTI3CqpqDvM2Avf9IDOQNeRq_xl.webp)
Binding of TRAIL homotrimer induces trimerization of DR4 and DR5, leading to the assembly of the death-inducing signalling complex (DISC) and subsequent recruitment of the adaptor protein Fas-associated death domain (FADD).[42] The latter acts as a bridge between the death receptor complex and the caspase-8 initiator prodomain. Upon activation of the extrinsic cell death pathway, dimerization of caspase-8 in the DISC leads to the formation of mature caspase-8, which activates immediate downstream effector caspases such as caspase-3, -6, and -7, triggering apoptosis.[43] The catalytically inactive caspase-8-homologous protein c-FLIP (FLICE-like inhibitory protein) can compete with caspase-8 for binding to FADD preventing active DISC formation and mediating cell resistance to apoptosis.[44][45]
TRAIL binding to DRs is critical for activation of the extrinsic apoptotic pathway, and downregulation of DR4 and DR5 is sufficient to render cancer cells resistant to TRAIL.[46] It has previously been shown that DR4 and DR5 can undergo spontaneous and ligand-mediated endocytosis and recycling independent of cancer cell sensitivity to TRAIL.[47] Disruption of receptor trafficking by dysfunctional cargo transporter proteins and nuclear translocation signalling proteins can inhibit translocation of DR from the trans-Golgi network (TGN) to the plasma membrane, resulting in reduced surface expression of DR.[46] In addition to localization on the plasma membrane, DR4 and DR5 are found in the cytoplasm and nucleus.[48] It has been shown that TRAIL can induce nuclear translocation and chromatin localization of DR4 and DR5 highlighting an additional role for surface-activated DRs via direct trafficking and signalling inside the nucleus.[49] Two functional nuclear localization signals have been identified in the DR5 receptor sequence, which, when mutated, together with knockdown of importin 1 by siRNA, prevented the nuclear localization of DR5, leading to increased DR5 expression on the cell surface and sensitization of HeLa and HepG2 cells to TRAIL.[50] Recent studies have identified specific tumour-promoting functions of nuclear DR5 as a regulator of let-7 maturation.[51]
In so-called type II cells, TRAIL can trigger the intrinsic apoptotic pathway, where activated caspase-8 cleaves the proapoptotic BH3-only BCL-2 family protein Bid. Truncated Bid (tBid), in turn, activates the proapoptotic proteins Bak and Bax, leading to mitochondrial outer membrane permeabilization (MOMP) and cytochrome C release.[52] Subsequently, the apoptotic protease activating factor 1 (APAF1) protein binds to cytochrome C and activates caspase-9. At the same time, during MOMP, a SMAC/DIABLO (second mitochondrial caspase activator) is released, which blocks the activity of inhibitors of apoptosis proteins (IAPs), thereby promoting apoptosis.[53] Active caspase-9 activates caspase-3, -6 and -7 in the same way as caspase-8. Anti-apoptotic members of the BCL-2 protein family, including BCL-2, BCL-xL, BCL-w, and MCL1, negatively regulate the TRAIL-mediated intrinsic death pathway by preventing MOMP.[54]
In addition to apoptosis, TRAIL death receptors can induce non-canonical signalling, leading to tumour resistance.[18][55] Activation of pro-inflammatory, pro-survival and proliferation pathways leading to pro-tumourigenic and, in some cases, metastasis-promoting effects following TRAIL treatment in vitro and in vivo have been described.[17][56-58] Non-canonical signalling is mediated by the formation of a secondary signalling complex that includes, in addition to FADD and caspase-8, RIPK1 (serine/threonine protein kinase 1), TRAF (TNF receptor-associated factor 2) and TRADD (TNF receptor-associated death domain).[56] This signalling complex can then activate various pro-tumourigenic pathways, including PI3K, MAPK/ERK, IκB/NF-κB, STAT3, Akt, Src and JAK2.
Thus, activation of non-canonical TRAIL pathways is a challenge not only because of the development of tumour resistance to DR agonists, but more importantly of the potential for tumour dissemination and metastasis.
A graphical representation of the TRAIL signalling pathways is shown in Fig. 2.
![[{"id":"uc0lWDiHck","type":"paragraph","data":{"text":"TRAIL signalling pathways. (<i>a</i>) Apoptosis pathway. Activation of DR4 and DR5 by TRAIL induces the extrinsic apoptosis pathway (left). The intrinsic pathway (right) is activated by a variety of stimuli and leads to the release of proapoptotic proteins from the mitochondria. The two pathways interact via caspase-8, which, once activated by the DRs, can cleave BID, further activating the intrinsic pathway, and conversely, caspase-3 can cleave and activate caspase-8 in a feedback loop, thus amplifying the apoptotic signal. (<i>b</i>) TRAIL-mediated non-apoptotic pathways. Anti- or pro-survival mechanisms appear to be context-dependent. DISC is death-inducing signaling complex; cFLIP is cellular FLICE [FADD-like IL-1β-converting enzyme]-inhibitory protein; SMAC is second mitochondrial activator of caspases; Cyt C is cytochrome C; XIAP is X-linked inhibitor of apoptosis. TRAF2 is TNF receptor-associated factor 2; NEMO is NF-kappa-B essential modulator; NF-κB is nuclear factor-κB; RIPK1 is receptor-interacting serine/threonine-protein kinase 1; JNK is C-Jun N-terminal kinase; ERK is extracellular signal regulated kinase, BAD is BCL-2-associated agonist of cell death; BIM is BCL-2-interacting mediator of cell death; PUMA is p53-upregulated modulator of apoptosis; NOXA is phorbol-12-myristate-13-acetate-induced protein 1; p38-BID is BH3-interacting domain death agonist; BAK is BCL-2/Killer 1 antagonist; BAX is BCL-2-associated X protein."}}]](/storage/images/resized/An61yKE65FsfzT95VEhtDlXDfflSMeDk3gHXqSYF_xl.webp)
3. Overcoming cancer cell resistance to TRAIL-induced apoptosis
Despite the high antitumour activity of TRAIL, it can also induce acquired resistance of tumour cells to apoptosis, which is considered an obstacle to its clinical use.[59] As a result, early clinical trials with several variants of TRAIL death receptor agonists have shown only limited therapeutic effect. Some of them showed insufficient antitumour activity, while for others, signs of toxicity were observed. Combination with various chemotherapeutic agents has not significantly improved the therapeutic potential of DR agonists, either because of a lack of appropriate combination therapy, or because of increased acquired cell resistance in response to the combination treatment.[21] It should be noted that many cancer cells are inherently resistant to TRAIL due to dysregulation of anti-apoptotic proteins such as BCL-2, BCL-XL, Bfl1/A1, c-FLIP, cIAP, survivin, Mcl-1 and XIAP.[18] To overcome TRAIL resistance, various chemotherapeutic agents and natural compounds have been proposed for combination therapy.[60] A number of synthetic small molecules have been obtained that suppress the activity of anti-apoptotic proteins and re-sensitize cancer cells to TRAIL.[22] To date, the most promising agents for combination therapy to overcome TRAIL resistance are proteasome inhibitors, BH3-mimetics, and cyclin-dependent kinase 9 (CDK9) inhibitors. Bortezomib is a selective 26S proteasome inhibitor approved by the US Food and Drug Administration (FDA) in 2003 under the trade name Velcade. It has demonstrated synergistic effects in various cancer cells when combined with DR agonists by increasing DR5 receptor expression and enhancing DISC formation.[61-63] Despite promising preclinical results, the combination of first-generation DR4 and DR5 agonistic antibodies, mapatumumab and conatumumab, correspondingly, with bortezomib failed to show additional therapeutic benefit (clinical trials NCT00315757, NCT00791011).
The BH3-only proteins initiate apoptosis by neutralizing the pro-survival BCL-2 family of proteins. Over the past 20 years, various small molecules called BH3-mimetics have been synthesized that mimic the function of BH3-only proteins and destroy cancer cells.[64] BH3-mimetics and TRAIL are highly synergistic in inducing apoptosis.[65][66] For example, a novel multimeric anti-DR5 IgM agonist antibody IGM-8444 (discussed further in Section 8) synergizes in vitro and in vivo with the BCL-2 selective inhibitor ABT-199 (also known as venetoclax, the only BH3 mimetic that has been approved by the FDA).[67]
Cyclin-dependent kinase 9 is a potential therapeutic target in cancer, as its overexpression correlates with cancer progression and poor clinical outcomes. CDK9 regulates multiple cellular functions including proliferation, survival, cell cycle regulation, DNA damage repair and metastasis. For this reason, many specific small molecule inhibitors have been developed over the past decades.[68] The novel, orally bioavailable CDK9 inhibitor Atuveciclib induced significant apoptosis in pancreatic cancer cell lines in combination with TRAIL via concomitant suppression of cFLIP and Mcl-1.[69] The clinically advanced CDK9-targeting drug Dinaciclib significantly enhanced the anticancer properties of TRAIL, regardless of the sensitivity or resistance of cancer cells to chemotherapy or targeted therapy.[70][71]
Dordaviprone (ONC201) is a first-in-class small molecule dopamine D2 receptor (DRD2) antagonist that upregulates TRAIL and death receptor 5 (DR5) expression by inactivating the pro-survival Akt/ERK kinases, thereby demonstrating potent antitumour activity in multiple cancer types.[72]
Recently, ONC201 monotherapy was shown to be well tolerated and to have long-term and clinically meaningful efficacy in recurrent diffuse midline glioma with H3 K27M mutation.[73] Other ONC201 analogues, designated ONC206 and ONC212, are under intensive in vivo and in vitro investigation, and have shown comparable or superior antitumour activity to ONC201.[74][75] The combination of imipridones ONC201 or ONC206 with temozolomide and radiotherapy also reduced intracranial tumour burden and prolonged survival in an orthotopic mouse model of IDH (isocitrate dehydrogenase) wild-type glioblastoma.[76]
Resistance of cancer cells to DR agonists may also be due to overexpression of members of the inhibitor of apoptosis protein (IAP) family. Drugs called SMAC mimetics have been developed that mimic natural IAP antagonists such as the second mitochondrial activator of caspase (SMAC).[77] The combination of such molecules with TRAIL overcomes cancer cell resistance either by degrading cIAP1 or XIAP, or by suppressing cFLIP(L).[78-80]
4. TRAIL fusions and conjugates aimed at increasing antitumour efficiency by improving protein stability and pharmacokinetic parameters
To overcome the aforementioned obstacles, several modification strategies have been developed to improve the antitumour properties of TRAIL death receptor agonists (FIg. 3).
![[{"id":"0KqcOjxMBG","type":"paragraph","data":{"text":"Schematic representation of fusion proteins targeting TRAIL death receptor pathway. Fusions of TRAIL with moieties for trimer stabilization, antitumour peptides, antibody fragments, or death receptor agonistic bispecific antibodies were designed to improve stability, pharmacokinetics, and antitumour efficacy."}}]](/storage/images/resized/gpwhy4F6dvyFVHSbHSXWY7qFtekj3JtujAJqPK4E_xl.webp)
Increasing the serum stability of TRAIL and prolonging its circulation in blood is a viable strategy to improve its antitumour efficacy. It is well known that the serum half-life of TRAIL is as low as 1 h.[8] Due to fast elimination from the body via renal clearance and poor pharmacokinetic profile, TRAIL signalling is insufficient to induce apoptosis in cancer cells. Unsatisfactory stability and pharmacodynamics have been among the challenges, which hampered the clinical translation of TRAIL-based therapies.[81] Recombinant soluble TRAIL can exist in different oligomeric states.[82][83] The hyper-oligomerized forms of TRAIL demonstrated extremely high potency in inducing apoptosis. Therefore, the oligomeric state of TRAIL can influence its stability and biological activity.[84]
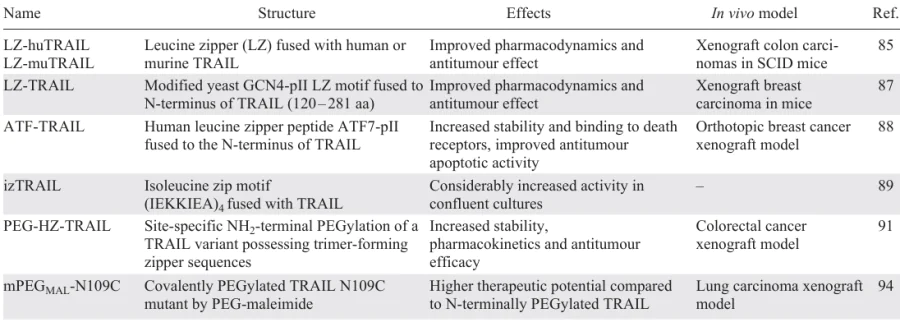
One of the first promising TRAIL modifications was leucine or isoleucine zipper-TRAIL.[85-89] The unique structural modification of zipper-TRAIL involves the introduction of hydrophobic interactions and intermolecular forces that promote self-assembly into stable multimeric structures.[88] These structures often adopt a ‘zipper-like’ pattern, where the TRAIL molecules align and bind together in a highly organized manner. Studies in breast, prostate and lung carcinomas have shown that self-assembly not only increases the stability of Zipper-TRAIL but also contributes to its increased resistance to proteolytic degradation. These stable structures not only protect Zipper-TRAIL from rapid degradation in the bloodstream, but also have the potential to enhance its binding to DRs on cancer cells, thereby promoting the apoptotic signalling.
A number of chemical modifications has been used to improve the therapeutic capacity of TRAIL. One such modification is the chemical attachment of polyethylene glycol (PEG). PEG is a non-immunogenic biological compound consisting of repeating units of ethylene glycol. Covalent and non-covalent attachment of PEG to proteins increases half-life, reduces immunogenicity and improves solubility and stability of therapeutic drugs.[90] According to this strategy, an N-terminally PEGylated TRAIL derivative (PEG-HZ-TRAIL) with trimer-forming zipper sequences has been constructed.[91] Site-specific PEGylation of the NH2 terminus of HZ-TRAIL with methoxypoly(ethylene glycol) aldehyde (mPEG-ALD) was carried out by reductive amination in the presence of sodium cyanoborohydride at acidic pH. Due to the relatively lower pKa values of the NH2-terminal amine (α-amino group) compared to that of the internal lysine residues (of e-amino groups), the reductive amination is quite selective for N-terminal – specific PEGylation. In vivo experiments in a colorectal carcinoma model showed increased stability of PEG-HZ-TRAIL due to its protection from proteolytic degradation in the bloodstream. PEGylation masks the TRAIL surface, reducing its vulnerability to enzymatic cleavage. In additions, the prolonged circulation and improved pharmacokinetics result in increased DR binding on cancer cells, leading to more efficient apoptotic signalling. Further, this PEGylated long-acting recombinant human TRAIL has been shown to ameliorate liver fibrosis and cirrhosis by selectively depleting activated hepatic stellate cells [92] and to induce DR-mediated apoptosis in collagen-producing myofibroblasts through upregulated DR5, reversing established skin fibrosis in scleroderma.[93]
In another work, site-specific PEGylation was performed using click chemistry approach by covalently binding PEG-maleimide to the TRAIL/N109C mutant bearing N-terminal cysteine residue. mPEGMAL-N109C showed improved in vitro stability and greater therapeutic potential in a tumour xenograft model, with better drug delivery and bioavailability compared to the TRAIL N-terminally PEGylated using mPEG-ALD.[94] The TRAIL/N109C variant was used to produce N109C-vcMMAE and PEG-TRAIL-vcMMAE conjugates with a cathepsin-cleavable linker between TRAIL and monomethyl auristatin E (MMAE).[95][96] MMAE is a potent antimitotic agent that inhibits cell division by blocking the polymerization of tubulin. Upon receptor binding, these fusions rapidly internalize into the cytoplasm of cancer cells and release MMAE into the lysosome via lysosomal-specific linker cleavage to induce growth arrest and cell death of cancer cells via apoptosis. Both fusions showed high antitumour activity and improved pharmacokinetic parameters. The half-life of PEG-N109C-vcMMAE in the rat was prolonged to 11.54 h compared to TRAIL/N109C (1.91 h) and N109C-vcMMAE (4.26 h).
One promising approach is to use trimerization domains derived from various proteins to enhance the stability of target proteins. These domains can be employed to promote the self-association of monomeric proteins into trimeric structures, which is often critical for the expression of their biological functions. The trimerization domain (TD) of human collagen, characterized by a repeating Gly-X-Y motif, imparts stability to proteins through its ability to self-assemble into a triple-helical structure that mimicks the structure of native collagen. TRAIL-TD fusion halved the IC50 in small-cell lung cancer cell model.[97] Another way to stabilize TRAIL is to fuse it to the Trimer-Tag, a human C-propeptide of alpha1(I) collagen. A recombinant fusion protein, SCB-313, consisted of Trimer-tag fused to the TRAIL C-terminus, allowing TRAIL to form a stable covalently linked homotrimer. SCB-313 had an extended half-life in the circulation due to its resistance to enzymatic cleavage and rapid clearance. The improved pharmacokinetic profile ensured sustained bioavailability, allowing for a longer therapeutic window and enhanced antitumour activity in colon carcinoma models in vitro and in vivo. The trimeric configuration not only stabilized the protein, but also increased its binding affinity to death receptors on cancer cells, resulting in more potent apoptotic signalling and increased cancer cell death.[98]
Another stabilizing modification of soluble TRAIL is fusion with an adenovirus knobless fibre motif. Three engineered TRAIL variants, designated FA1FT, HA5FT and HA5ST, were created by fusing the avian Ad1 spineless fibre (FA1FT), the N-terminal tail and the first two repeats of the human Ad5 fibre shaft (HA5FT) and the last repeat of the human Ad5 fibre shaft (HA5ST) to the N-terminus of TRAIL. These constructs were designed to form a trimeric configuration in which the three TRAIL monomers are linked to each other via the adenovirus fibre rod domain. Among these proteins, HA5ST showed the highest anticancer activity and improved pharmacodynamics.[99]
It has also been shown that improved TRAIL stability can be achieved by genetically fusing TRAIL to the albumin binding domain (ABD).[100] The ABD component of the ABD-hTRAIL fusion promoted binding to albumin in the bloodstream, which significantly prolonged the half-life, making ABD-hTRAIL more stable than native TRAIL. The presence of ABD protected TRAIL from proteolytic degradation, reducing its susceptibility to enzymatic cleavage and denaturation. This protein has been tested in vivo in models of colon carcinoma and has shown a remarkable ability to induce cancer cell death, resulting in tumour growth inhibition. The same group of researchers created a fusion in which the IgG-binding affibody, IgBD, was genetically fused to the N-terminus of TRAIL to form IgBD-TRAIL, and compared its cytotoxicity, serum half-life, and antitumour activity with ABD-TRAIL.[101] The serum half-life of both fusions exceeded that of TRAIL but the antitumour effects of the intravenously injected IgBD-TRAIL were superior to that of ABD-TRAIL.
Aiming to improve the TRAIL stability, another research group[102] has fused the crystallizable region of a human IgG1 fragment (Fc) with the TRAIL N-terminus to produce an Fc-TRAIL fusion. The chimeric protein had a significantly longer half-life in mice and was more effective than TRAIL in inhibiting tumour growth in a xenograft model of lung cancer.
A strategy combining increased stability and additional antitumour activity was also implemented by fusing TRAIL to a hexameric arginine deiminase (ADI). ADI-TRAIL benefited from the structural and functional synergy between the two moieties and had an extended half-life in vivo. It not only activated the DR5 receptor, but also suppressed survivin, and sensitized cancer cells to TRAIL-induced apoptosis significantly inhibiting tumour growth in colorectal carcinoma models.[103]
An interesting modification of recombinant mutant human TRAIL (rmh TRAIL) developed by Beijing Sunbio Biotech and further renamed CPT (circularly permuted TRAIL, drug name Aponermin) consists of the N-terminal TRAIL amino acids (121 – 135) fused with a flexible linker Gly5 to the C-terminal TRAIL amino acids (135 – 281).[104] CPT has potent antitumour activity in vitro and in vivo, and has demonstrated the clinical activity in a Phase II trial in patients with relapsed or refractory multiple myeloma as a single agent [105] and in a Phase III trial in combination with thalidomide and dexamethasone.[106]
To optimize the oligomerization, a Flag tag and chicken tenascin-C (TNC) oligomerization domain (110 – 139 amino acids) were fused to the N-terminus of TRAIL to obtain Flag-TNC-TRAIL.[107] Introduction of the TNC domain enhanced TRAIL activity after secondary cross-linking: oligomerized Flag-TNC-TRAIL strongly induced cell death in myeloma cells, but also activated pro-inflammatory signalling pathways.[108] An alternative way to force trimerization is a covalently linked TRAIL trimer (TR3) created by genetically fusing three consecutive extracellular domains of TRAIL in a head-to-tail configuration. This molecular design provided improved stability without altering the native killing capacity of TRAIL. The authors also generated an scFv-TR3 fusion of a single-chain antibody fragment (scFv) specific for murine red blood cells (RBCs) with the NH2-terminus of TR3. Similarly, scFv-S-TR3 variant with an elongated spacer domain between the targeting scFv and TR3 comprising four globular domains of the human complement regulatory proteins decay accelerating factor (DAF, CD55) and complement receptor 1 (CR1, CD35) was produced to anticipate the possible steric constraints. RBCs coated with the scFv-S-TR3 have demonstrated high potential for clinical cancer therapy in pancreatic adenocarcinoma models.[109]
A TRAIL-Mu3 fusion protein was obtained to enhance the membrane permeability of TRAIL by replacing the amino acid residues VRERGPQR (114 – 121) with RRRRRRRR.[110][111] TRAIL-Mu3 more effectively activated the caspase cascade in pancreatic cancer cell lines and demonstrated increased efficacy in preclinical models of pancreatic cancer compared to wild-type TRAIL (wtTRAIL).
Another original approach was used to generate biologically functional DR4 and DR5 agonists (KD413, KD 506 and KD548) from a Kringle domain (KD) scaffold by generating a synthetic KD library on the surface of yeast cells and randomizing 45 residues in the loops of the human KD template.[112] The KD variants selected against DR4 and DR5 showed antitumour activities in vitro and in vivo. In addition, using a loop grafting technique, the authors created a bispecific kringle domain agonist bvKD548-55 as a potent agonist of death receptors 4 and 5.[113]
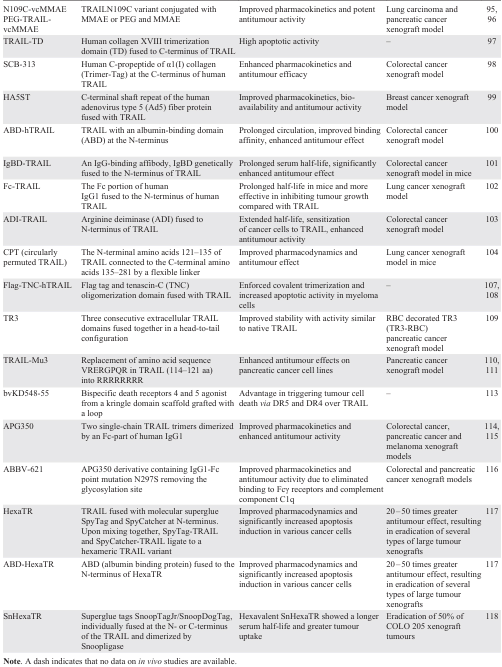
5. Fusions aimed at increased clustering of the death receptors DR4 and DR5
One of the reasons for the limited efficacy exhibited by recombinant soluble trimeric TRAIL is its insufficient receptor clustering capacity during short-term presence in the bloodstream. Since the efficacy of DR agonists in inducing tumour cell apoptosis is highly dependent on their valency, therefore, recent efforts have focused on improving DR clustering to enhance antitumour activity. A potent TRAIL receptor agonist APG350 with a hexavalent binding pattern that allows clustering of six TRAIL receptors per drug molecule, was produced by C-terminal fusion of an engineered IgG1-Fc to a single-chain TRAIL receptor binding domain (scTRAIL-RBD).[114] Antitumour efficacy of APG350 exceeded that of soluble TRAIL in pancreatic cancer with limited recurrent tumour growth and metastases.[115] Subsequently, an APG350 fusion derivative ABBV-621 was created containing a single IgG1-Fc point mutation (asparagine 297 to serine) that effectively removes the glycosylation site to eliminate binding to all Fcγ receptors and complement component C1q. Preclinical studies have demonstrated improved pharmacokinetics and antitumour activity of ABBV-621, particularly when combined with chemotherapeutic agents such as docetaxel, irinotecan, and the selective BCL-XL inhibitor A-1331852, in preclinical models of colorectal and pancreatic cancer.[116]
Novel genetic engineering techniques using small molecule superglues to covalently ligate proteins offer promising alternatives for protein oligomerization. Using the molecular superglue SpyTag/SpyCatcher to create more complex TRAIL variants, such as hexameric TRAIL (HexaTR), resulted in a significant increase in apoptosis induction.[117] The development of albumin-binding HexaTR (ABD-HexaTR) with an extended serum half-life demonstrated its remarkable antitumour activity in vivo, effectively eliminating various xenograft tumours. These data suggest that the superglue-mediated higher-order assembly approach is valuable for improving pro-apoptotic TRAIL signalling and has significant potential for advancing DR agonists in cancer therapy. The same research group [118] further generated hexavalent fusion SnHexaTR by catalyzing trivalent TRAIL variants with N-terminal fusion of SnoopTagJr/SnoopDogTag with Snoopligase. The in vitro cytotoxicity of SnHexaTR was 10 – 40 times greater than that of TRAIL in several tumour cell lines. SnHexaTR showed a longer serum half-life and greater tumour uptake than TRAIL, resulting in enhanced cytotoxicity and antitumour activity of TRAIL.
A summary of TRAIL fusions aimed at improving protein stability and pharmacokinetic parameters is presented in Table 1.
6. Bifunctional fusions of TRAIL with antitumour peptides or proteins
Genetically engineered bispecific proteins integrate functionally distinct protein fragments into a single molecule and thus exert diverse biological effects, such as acting as a drug or drug transporter, or both.[119] Fusions of TRAIL with antitumour peptides or proteins have shown promising potential to enhance its efficacy in cancer therapy. The conjugation of TRAIL with specific peptides or proteins can lead to improved cell recognition, targeted tumour delivery, and enhanced apoptotic signalling. As a part of these molecular constructs, TRAIL is able to selectively engage its cognate DRs on cancer cells, triggering the extrinsic apoptosis pathway while minimizing off-target effects on healthy tissues.[120] This strategy not only enhances the apoptotic response but also overcomes resistance mechanisms commonly found in cancer cells, ultimately increasing the overall therapeutic impact of TRAIL-based treatment in various malignancies.
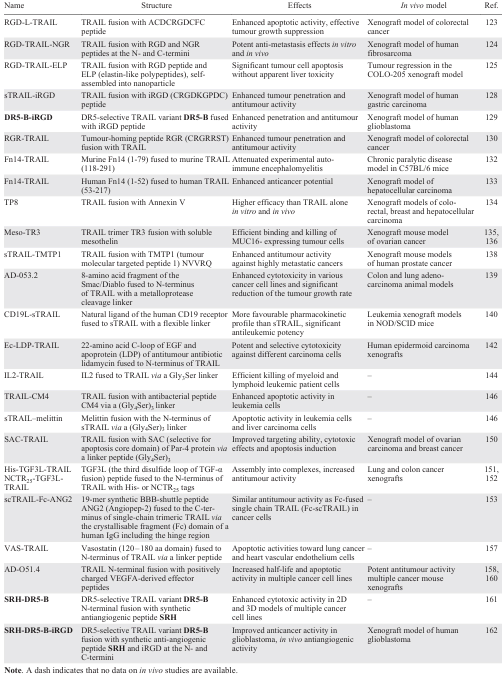
Integrins ανβ3 and ανβ5 play crucial roles in various aspects of cancer progression and metastasis. Their upregulation is associated with tumour growth, angiogenesis and invasion, making them attractive targets for cancer therapy. Targeting integrins ανβ3 and ανβ5 has been explored as a potential strategy to inhibit cancer progression and metastasis, making them important targets for the development of anticancer therapeutics.[121][122] In particular, RGD (Arg-Gly-Asp) peptides specifically target the integrins ανβ3 and ανβ5, which are commonly overexpressed in both the tumour neovasculature and tumour cells. RGD-L-TRAIL fusion protein combines the peptide ACDCRGDCFC, which contains the RGD cell adhesion motif, with TRAIL to enable double-targeted delivery of TRAIL to the tumour site. RGD-L-TRAIL showed promising results in inhibiting the proliferation of various cancer cell lines and significantly inhibiting tumour growth in mice with colon cancer tumour xenografts.[123] Another fusion RGD-TRAIL-NGR, containing the tumour-targeting peptides RGD and NGR at the N- and C-terminus of TRAIL, respectively, demonstrated improved anticancer efficacy and metastasis inhibition activity.[124] Another fusion protein, RGD-TRAIL-ELP, was developed by fusing the C-terminus of RGD-TRAIL with elastin-like polypeptides (ELPs), which were able to self-assemble into nanoparticles under physiological conditions. RGD-TRAIL-ELP demonstrated an enhanced apoptosis-inducing ability, and a single intraperitoneal injection of nanoparticles resulted in almost complete tumour regression in the COLO-205 tumour xenograft model.[125]
RGD peptides can be either linear or cyclic. Cyclic RGD peptides have higher activity than linear RGD peptides due to their more stable conformation that resists proteolysis.[126] The cyclic iRGD peptide (CRGDKGPDC) exploits the tumour microenvironment by activating the integrin-dependent binding to the tumour vasculature, and neuropilin-1 (NRP-1)-dependent transport into tumour tissues.[127] A sTRAIL-iRGD fusion was developed to address the limitations of traditional TRAIL therapy, such as low tumour permeability and limited efficacy. The sTRAIL-iRGD achieved improved tumour-specific delivery, more effective apoptosis-inducing capacity compared to TRAIL, and exhibited antitumour effects more effectively in a gastric cancer model.[128] Another fusion peptide DR5-B-iRGD was generated by fusing iRGD to the C-terminus of the DR5-selective TRAIL mutant variant DR5-B.[129] DR5-B-iRGD penetrated into U87 tumour spheroids faster than DR5-B, demonstrated an enhanced antitumour effect in human glioblastoma cells and more effectively inhibited tumour growth in a xenograft mouse model of human glioblastoma.
The fusion of the tumour-homing RGR peptide (CRGRRST) with TRAIL to produce RGR-TRAIL has been reported.[130] RGR-TRAIL showed enhanced cell binding and cytotoxicity in colorectal cancer (CRC) cells compared to TRAIL, and exerted significantly enhanced growth suppression in mice bearing CRC tumour xenografts. The in vitro and in vivo antitumour effects of RGR-TRAIL were significantly improved by combination with (EGFR)-targeted photodynamic therapy (PDT).
The Fn14 (fibroblast growth factor-inducible 14-kDa protein) signalling axis, which is activated by its ligand TWEAK (TNF-like weak inducer of apoptosis), is involved in tumour cell proliferation through autocrine and paracrine signalling and can promote cancer progression through pro-survival and pro-angiogenic effects.[131] An Fn14-TRAIL fusion containing 1 – 79 amino acid fragment of the murine Fn14 receptor with murine TRAIL (118 – 291) was generated to attenuate an experimental autoimmune encephalomyelitis.[132] Later, the similar Fn14-TRAIL fusion of the extracellular domain of human Fn14 (1 – 52) with human TRAIL (53 – 217) demonstrated higher apoptotic activity than TRAIL alone in hepatocellular carcinoma cell lines and strong anticancer properties in vivo.[133]
Annexin V, a member of the Annexin superfamily, is known for its involvement in various cellular processes such as blood coagulation, signal transduction, and anti-inflammatory responses. In the context of cancer therapy, the development of a chimeric protein Annexin V-TRAIL (TP8) has shown promising results in the selective induction of apoptosis in various tumour cell types, both in vitro and in vivo.[134] This fusion protein exhibited superior efficacy compared to native TRAIL representing a potential strategy for the treatment of TRAIL-resistant cancers.
MUC16 (CA125), a well-established biomarker in ovarian cancer, is also highly expressed in other cancers including pancreatic and breast cancers. Meso-TR3, a fusion of TR3 (Ref. [109]) with mesothelin, which specifically binds MUC16, is a novel therapeutic that induces potent cell death by targeting via an additional tumour-specific moiety. Meso-TR3 has been shown to have a significantly higher killing capacity on MUC16-expressing cancer cells compared to non-targeted TR3 and recombinant TRAIL.[135] The specificity of Meso-TR3 for MUC16-positive cells was validated in various in vitro and in vivo experiments, assuming its potential for homotypic (tumour cell-tumour cell) and heterotypic (tumour cell-mesothelial cell) cell interactions.[136]
The synthetic 5-amino acid peptide TMTP1 (NVVRQ) identified by the FliTrx bacterial peptide display system specifically binds highly metastatic tumour cells, including prostate, breast, lung and gastric cancers in vitro and in vivo, but not the non-metastatic cell lines.[137] In line with that, the sTRAIL-TMTP1 fusion protein demonstrated significant cytotoxic effects on highly metastatic tumour cells through the activation of the extrinsic apoptotic pathway. In preclinical studies, sTRAIL-TMTP1 effectively inhibited primary tumour growth and metastasis in various cancer models, and also showed a stronger suppressive effect on angiogenesis compared to standard TRAIL treatment.[138]
Second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (SMAC/DIABLO) is a pro-apoptotic mitochondrial protein that is released into the cytosol in response to various apoptotic stimuli and antagonizes inhibitors of apoptosis proteins (IAPs), thus allowing the activation of caspases and apoptosis. AD-053.2 is a fusion protein in which an 8-amino acid fragment of the SMAC/Diablo protein has been fused to the N-terminus of TRAIL with a metalloprotease cleavage linker.[139] The AD-053.2 protein showed a potent cytotoxic activity with the IC50 values in femtomolar range for the most sensitive cancer cell lines and significantly reduced the rate of the tumour growth in colon and lung adenocarcinoma animal models.
CD19, a B-lineage restricted receptor expressed on leukemia cells, represents a promising target for biotherapy in relapsed acute lymphoblastic leukemia due to its absence in critical non-hematopoietic organs and its abundant expression on relapsed rare mature B-cell leukemia clones. The recombinant human CD19L-sTRAIL fusion protein exhibited selective binding and potent apoptotic activity against CD19-positive leukemia cells, activating the TRAIL death pathway in a caspase 8-dependent manner. In preclinical studies, CD19L-sTRAIL demonstrated significant antileukemic potency against primary leukemia cells and in acute myeloid leukemia (AML) xenograft models without significant toxicity in animal models.[140][141] The favourable pharmacokinetic profile of the protein and efficacy in chemotherapy-resistant cases suggest its potential as a biotherapy for relapsed and refractory AML patients.
Lidamycin (LDM), an antitumour antibiotic, has potent cytotoxicity against cancer cells in vitro and in vivo.[141] The bifunctional Ec-LDP-TRAIL fusion protein combines the EGF C-loop (22 amino acids of the EGF C-terminus), the Lidamycin apoprotein (LDP) and the functional TRAIL. In addition, the energized fusion protein Ec-LDP-TRAIL-AE was created by mixing Ec-LDP-TRAIL with enediyne chromophore AE. Ec-LDP-TRAIL-AE exhibited specific binding to EGFR and TRAIL DRs and induction of apoptosis in tumour cells expressing these receptors. In both in vitro and in vivo studies, Ec-LDP-TRAIL-AE showed enhanced antitumour activity compared to LDM alone, while sparing normal organs such as the liver, lung, and intestine in experimental animals.[142]
The cytokine IL2 has been approved for the treatment of metastatic renal cell carcinoma and metastatic melanoma. It has been successfully used to combat various malignant neoplasms due to its ability to induce the proliferation of natural killer cells, T cells and B cells, thereby enhancing cellular immunity against cancer.[143] The IL2-TRAIL fusion showed higher cytotoxicity in leukemic cell lines compared to TRAIL. This immunotoxin was found to be highly specific in targeting CD25-positive leukemia cells, showing the potential as a potent and selective therapeutic agent.[144]
Antimicrobial peptides (AMPs) with high efficacy and low toxicity are promising new drugs that can replace chemoradiotherapy.[145] The antibacterial peptide CM4 is a cationic linear α-helical 35-amino acid peptide, which belongs to the cecropin family. The most intriguing feature of CM4 is its destructive effect on bacteria, fungi and tumours without damaging normal cells. TRAIL-CM4, a fusion protein of TRAIL and CM4, has shown potent antitumour activity, inducing higher rates of cell death and apoptosis in cancer cells compared to TRAIL alone.[146]
sTRAIL-melittin [147] is another TRAIL fusion with a 26-amino acid antibacterial peptide melittin with anticancer activity, derived from bee venom.[148] Despite its potential to induce tumour cell death through multiple pathways, the non-selective toxicity and side effects of melittin restrict its application as a viable anticancer agent in vivo. sTRAIL-melittin fusion displayed enhanced anticancer activity against leukemia and liver cancer cells, inducing significant apoptosis, while demonstrating no cytotoxic effects on normal erythrocytes and human embryonic kidney cells,[147] highlighting its potential for cancer therapy.
The selective apoptosis activator Par-4 (prostate apoptosis response-4) is a multi-domain protein containing the 59-amino acid SAC (selective for apoptosis of cancer cells) domain, which enables it to specifically induce apoptosis in cancer cells without harming normal cells or tissues.[149] Fusing the Par-4 SAC domain with TRAIL resulted in enhanced anticancer potential of the resulting SAC-TRAIL fusion, creating a promising candidate for cancer therapy. The structure and sequence order of the fusion protein were optimized using various flexible linkers for expression in E. coli.[150]
Enhancement of the anticancer effect of TRAIL was demonstrated by a TGF3L-TRAIL fusion, in which a synthetic TGF3L (the third disulfide loop of TGF-α) peptide was linearized to eliminate any possible residual EGFR binding affinity and fused to the N-terminus of TRAIL.[151] The TGF3L-TRAIL fusion showed increased cytotoxicity in a variety of cancer cell lines in vitro and in vivo. The authors showed that TGF3L-TRAIL forms stable polymers, which provides increased cytotoxicity against cancer cells. Importantly, the TGF3L-TRAIL fusion was designed with a His tag at the N-terminus. In another attempt, the truncated sequence from the N-terminus of hCtr1 (human copper transporter 1) (NCTR25-tag) was used instead of the classic His-tag, which is not well suited for clinical applications to produce TRAIL in E. coli.[151, 152] Both NCTR25-TRAIL and NCTR25-TGF3L-TRAIL demonstrated the ability to self-organize into a polymer-like structures, and exhibited significantly higher activity compared to TRAIL via selective activation of DR4 and DR5 receptors.
To improve the penetration of TRAIL molecules across the blood-brain barrier (BBB), the small 19-amino acid peptide ANG2 (Angiopep-2) was fused to single-chain scTRAIL via the crystallizable fragment (Fc) domain of a human IgG including the hinge region for the treatment of glioblastoma (Fig. 4).[153] The Angiopep family of peptides are derived from the Kunitz domain of human aprotinin, which can cross the BBB and are used to facilitate the delivery of pharmacological agents to the brain. The Angiopep-2 has demonstrated the ability to facilitate LRP1 (low-density lipoprotein receptor-related protein 1)-dependent transcytosis across the BBB and has been used to enhance the CNS (central nervous system) penetrance of various cargoes, including drugs, proteins and nanoparticle-based systems, with early clinical trials showing promisingly low toxicity.[154] The hexavalent scTRAIL-Fc-ANG2 fusion protein remained highly effective in inducing apoptosis in glioblastoma cells, but binding to BBB cells was predominantly due to the TRAIL protein as TRAIL has a significantly higher binding rate for its receptors than ANG2 to the LRP1 receptor.[153]
![[{"id":"YmOvRGzh9G","type":"paragraph","data":{"text":"Design of a CNS-targeted TRAIL-receptor agonist. (<i>a</i>) Functional units, (<i>b</i>) composition and (<i>c</i>) schematic assembly of CNS-targeted scTRAIL variants and relevant control proteins. Reproduced from Moorthy et al.[[ type=\"anchor\" referenceId=\"13985\" ]] under the Creative Commons Attribution (CC BY) license https://creativecommons.org/licenses/by/4.0/)."}}]](/storage/images/resized/8Vv1SDMDWIPd0bGNphIi7kXHGdlaR670hsq7KoJL_xl.webp)
The formation of new blood vessels (angiogenesis) is a complex and dynamic process regulated by various pro- and antiangiogenic molecules. Angiogenesis plays a key role in tumour growth, invasion and metastasis.[155] Treatment of tumour diseases with antiangiogenic agents is a promising strategy for antitumour therapy. However, the use of these drugs is still limited due to several disadvantages such as side effects, acquired drug resistance and tumour recurrence. In order to simultaneously target the tumour and tumour microenvironment, several TRAIL fusions with antiangiogenic peptides have been developed.
Vasostatin is a naturally occurring endogenous peptide derived from the cleavage of calreticulin, a multifunctional protein involved in several cellular processes. This peptide has been implicated in the regulation of angiogenesis, the process by which new blood vessels are formed from the pre-existing ones.[156] VAS-TRAIL, a fusion protein combining vasostatin and TRAIL moieties, inhibited tumour cell growth and induced apoptosis in cancer cells, while significantly suppressing proliferation in fetal bovine heart endothelial (FBHE) cells. The combined anti-angiogenic properties of vasostatin and apoptotic effects of TRAIL resulted in increased endothelial cell apoptosis compared to either vasostatin or sTRAIL alone, demonstrating the effective integration of both domains within the VAS-TRAIL fusion protein.[157]
Another fusion protein, AD-O51.4, is composed of a TRAIL-derived sequence linked to VEGFA-derived peptides. AD-O51.4 engages a multifaceted mechanism, involving both extrinsic and intrinsic apoptotic pathways, and triggers potent inhibition of tumour growth. Its potential to induce apoptosis in both tumour cells and endothelial cells highlights the importance of targeting the tumour microenvironment for effective therapeutic intervention.[158]
AD-O51.4 reduced the growth of CRC PDXs (patient-derived xenografts) with good efficacy and displayed cytotoxic effects in DLBCL (diffuse large B-cell lymphoma) cells through induction of DR-mediated caspase-dependent apoptosis.[159, 160]
We have also recently generated TRAIL-based fusions SRH-DR5-B [161] and SRH-DR5-B-iRGD [162] with antiangiogenic synthetic peptide SRH (SRHTKQRHTALH) at the N-terminus of DR5-selective TRAIL mutant variant DR5-B [33] or DR5-B-iRGD [129] via flexible linker (Fig. 5). DR5-B selectively binds the DR5 receptor and induces apoptosis more potently than TRAIL.[13, 61, 163] Both DR5-B-based fusions demonstrated enhanced antitumour activity in vitro and in vivo compared to DR5-B. Importantly, they internalized the DR5 receptor faster than DR5-B and exhibited antiangiogenic activity in glioblastoma xenografts, with these effects being more pronounced for SRH-DR5-B-iRGD due to the additional αvβ3 integrin-specific iRGD peptide moiety.
![[{"id":"BSjsguY1jQ","type":"paragraph","data":{"text":"Fusion of DR5-selective TRAIL mutant variant DR5-B with antiangiogenic synthetic peptide SRH (SRHTKQRHTALH) and cell-penetrating peptide iRGD (CRGDKGPDC) for dual targeting of VEGFR2 and integrin αvβ3 receptors."}}]](/storage/images/resized/XhWinmwGIlubfzl4ruA83ZXLjd7y9NDEcj5ugeQ5_xl.webp)
A summary of TRAIL-based bifunctional fusions is presented in Table 3.
7. TRAIL fusions with antibody fragments targeting tumour-specific antigens
Single-chain fragment variable (scFv) antibodies have become valuable tools in cancer therapy due to their ability to specifically target tumour-associated antigens. These are single 25 kDa polypeptides that contain the variable light chain (VL) and the variable heavy chain (VH) regions of the antibody connected by a flexible linker peptide, which is usually 15 – 20 amino acids long and consists of glycine and serine with dispersed hydrophilic residues for increased solubility.[164, 165] Single-chain variable fragments, which contain the complete antigen-binding domains of the whole antibody, have several advantages, such as high specificity and affinity for antigens, low immunogenicity, and the ability to penetrate and diffuse into tumour tissue.[166]
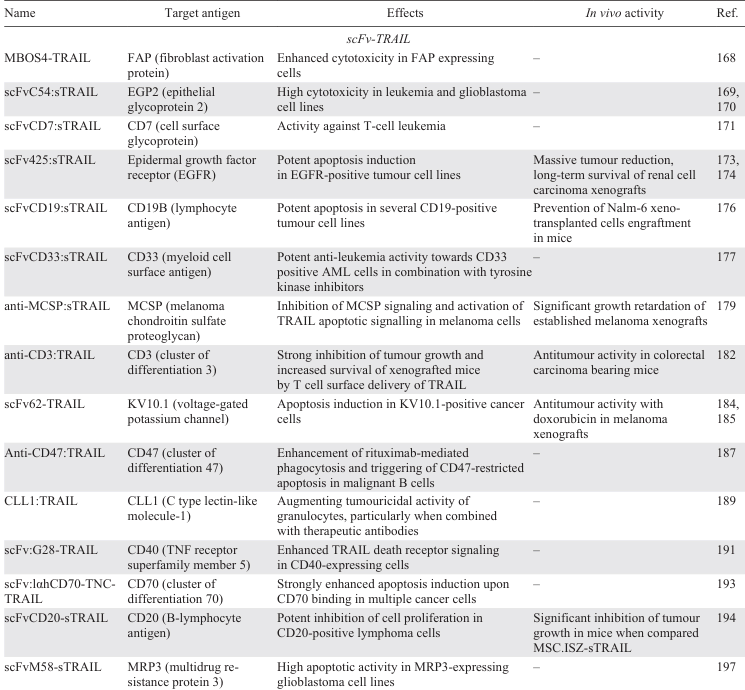
Over the past decades, numerous TRAIL or single-chain TRAIL (scTRAIL) fusions with scFc antibodies against various target surface antigens known to be highly expressed by certain tumour types have been generated to enhance antitumour activity and improve pharmacokinetic parameters.[19, 20, 167]
The first scFv-Fc fusion, MBOS4-TRAIL, was generated in 2001 by fusing a recombinant tumour stroma marker FAP (fibroblast activation protein)-specific single-chain antibody fragment MBOS4 and the Fc fragment of human IgG to the extracellular domain of TRAIL via linkers. Dose-response analysis of MBOS4-TRAIL-induced apoptosis revealed approximately 30-fold higher toxicity towards FAP-expressing cells compared to TRAIL.[168]
Two fusions with antitumour bystander activity, scFvC54:sTRAIL and scFvCD7:sTRAIL, were generated by fusing the scFv fragments specific for epithelial glycoprotein 2 (C54) and cell surface glycoprotein (CD7) to the N-terminus of human sTRAIL. The scFvC54:sTRAIL showed favourable properties, potentially reducing the amount of sTRAIL required for antitumour activity.[169, 170] The scFvCD7:sTRAIL, which contains an scFv antibody fragment, specific for human CD7, which is abundantly expressed on most T cell malignancies and 10% of acute myeloid leukemias, showed enhanced and target antigen-restricted apoptotic activity against human T cell acute lymphoblastic leukemia (T-ALL) cells without toxicity to normal human blood and endothelial cells. In mixed culture experiments with CD7-positive and CD7-negative tumour cells, scFvCD7:sTRAIL induced very potent bystander apoptosis.[171]
The EGFR signalling plays a key role in the regulation of various tumour cell functions such as cell cycle progression, inhibition of apoptosis, angiogenesis, cell motility, adhesion, and invasion.[172] An engineered EGFR-specific scFv425:sTRAIL fusion protein constructed of a fragment of the EGFR-blocking antibody scFv425 genetically fused with soluble TRAIL, demonstrated a significant apoptosis-inducing activity in a number of EGFR-positive tumour cell lines.[173] Treatment of mice bearing intraperitoneal renal cell carcinoma xenografts with the adenovirus-expressed Ad-scFv425:sTRAIL fusion resulted in a rapid and massive reduction in tumour burden and subsequent long-term survival.[174]
A pan-B cell marker CD19 has been recognized as a potential immunotherapy target for B cell disorders, including blood-borne malignancies and autoimmune diseases.[175] The scFvCD19:sTRAIL fusion protein demonstrated CD19-specific apoptosis induction in B-ALL cell lines, while sparing normal hematopoietic cells. It also showed bystander apoptosis-inducing effects in CD19-negative tumour cells and significant therapeutic potential in inhibiting tumour engraftment and prolonging survival in mice xenografted with human B-ALL cells.[176]
Another antigen-specific antibody fragment against CD33 was used to generate the scFvCD33:sTRAIL fusion.[177] CD33 is a cell surface antigen expressed in ~ 80 – 90% of AML patients, particularly on leukemic blasts, making it a target for therapy.[178] Ex vivo treatment of patient-derived CD33-positive AML cells with scFvCD33:sTRAIL resulted in potent induction of apoptosis. In chronic myeloid leukemia (CML) cells, scFvCD33:sTRAIL had potent antileukemic activity against CD33+ cells when combined with the Bcr-Abl tyrosine kinase inhibitor, imatinib mesylate (Gleevec).[177]
The anti-MCSP:sTRAIL fusion protein designed to target melanoma cells with overexpressed CSPG4 (chondroitin sulfate proteoglycan 4), demonstrated potent apoptosis induction and tumour growth retardation in preclinical models.[179] CSPG4 is overexpressed in several types of cancer, including breast cancer, melanoma, squamous cell carcinoma, mesothelioma, neuroblastoma, adult and pediatric sarcomas, and some hematological cancers. CSPG4 has been the target of numerous anticancer therapies aimed at inhibiting its signalling pathways that promote proliferation, migration, and invasion.[180] Targeting MCSP with anti-MCSP:TRAIL inhibited MCSP signalling and activated TRAIL apoptotic signalling in melanoma cells in vitro and in vivo.[181]
To augment the tumouricidal activity of T cells, two recombinant fusion proteins, anti-CD3:TRAIL and K12:TRAIL, were generated that selectively bind to the surface of T cells with CD3 and CD7 antigens, respectively. Anti-CD3:TRAIL contains a CD3 stimulatory antibody fragment, whereas K12:TRAIL contains a soluble form of the CD7 ligand K12. Both fusions strongly enhanced the tumouricidal activity of T cells against a panel of cancer cell lines, primary patient-derived malignant cells, and in a murine xenograft model.[182]
The voltage-gated potassium channel Kv10.1, a tumour-specific marker, has been recognized as a promising target in cancer therapy due to its high expression in tumour tissues.[183] A KV10.1-specific scFv antibody fused with sTRAIL has been developed for combination therapies to target prostate cancer cells with high tumour specificity.[184] The scFv62-TRAIL construct induced apoptosis in human prostate cancer cells after sensitization with cytotoxic drugs only in KV10.1-positive cancer cells, but not in non-transformed cells. The antitumour effect of scFv62-TRAIL in combination with doxorubicin was demonstrated in SCID mice bearing subcutaneous melanomas.[185]
CD47 is a key ‘don’t eat me’ signalling molecule that allows cancer cells to evade phagocytic clearance. Binding of CD47 to a SIRPα receptor on the surface of phagocytes inhibits the cancer cell clearance. Blocking the CD47-SIRPα interaction can enhance phagocytic clearance of cancer cells, making it a potential therapeutic approach to boost the efficacy of anticancer antibodies and promote apoptosis in malignant cells.[186] Anti-CD47:TRAIL fusion enhanced rituximab (RTX)-mediated phagocytosis of B cell non-Hodgkin’s lymphoma (B-NHL) cells and triggered apoptosis in CD47+ B cell lines and primary malignant B-NHL samples while sparing normal blood cells.[187]
Selective expression of C-type lectin-like molecule-1 (CLL1) on granulocytes, monocytes, and dendritic cells makes it a promising target for enhancing the antitumour activity of granulocytes for the AML therapy.[188] CLL1:TRAIL is a fusion protein designed to enhance the ability of leukocytes, primarily granulocytes, to significantly enhance their antitumour activity and increase the cytotoxicity of therapeutic anticancer antibodies. CLL1:TRAIL improved the overall efficacy of anti-cancer antibodies such as rituximab, cetuximab, alemtuzumab and daratumumab-based antibodies.[189]
Based on the Flag-TNC-hTRAIL fusion,[108] two novel proteins, scFv:G28-TRAIL and scFv:lαhCD70-TNC-TRAIL, bearing CD40- and CD70-specific scFv fragments have been constructed. CD40, a member of the TNF receptor superfamily, has shown promise in cancer immunotherapy by promoting antitumour immune responses when activated. Clinical trials using CD40 agonist antibodies, often in combination with other treatments such as checkpoint inhibitors and chemotherapy, have demonstrated encouraging antitumour effects, making CD40 activation a valuable strategy for improving cancer treatment.[190] CD40-dependent enhancement of apoptosis by scFv:G28-TRAIL has been demonstrated in various cell lines, providing potential for CD40-restricted cancer therapy.[191] Another member of the TNF family, CD70, interacts with the CD27 receptor to promote the expression of anti-apoptotic genes that boost T cell survival and proliferation.[192] The scFv:lαhCD70-TNC-TRAIL fusion, a stabilized form of TRAIL containing a llama CD70-specific scFv, demonstrated enhanced induction of apoptosis upon CD70 binding through effective interference with the CD70-CD27 interaction. scFv:lahCD70-TNC-TRAIL fusions containing DR4- and DR5-selective TRAIL mutant variants also enhanced the induction of cell death upon CD70 binding, with a preference for the DR5-selective variant.[193]
The scFvCD20-sTRAIL fusion was designed and expressed in human umbilical cord mesenchymal stem cells as a carrier for dual targeted therapy against non-Hodgkin’s lymphoma.[194] The expression of non-glycosylated CD20 protein has been detected on the surface of normal and malignant B cells and is considered an ideal therapeutic target, as rituximab-based immunotherapy has become the standard of care for most B-cell malignancies.[195] The scFvCD20-sTRAIL fusion protein demonstrated significant enhancement of cellular apoptosis through both extrinsic and intrinsic apoptotic signalling pathways. In the NOD/SCID mouse BJAB subcutaneous lymphoma xenograft model, intravenous injection of MSC.scFvCD20-sTRAIL significantly inhibited tumour growth compared to MSC.ISZ-sTRAIL-treated mice.[194]
MRP3, a multidrug resistance protein, has been identified as a specific antigen in glioblastoma multiforme (GBM), making it a valuable target for antibody-based cancer therapy.[196] A scFvM58-sTRAIL fusion protein, designed to selectively induce apoptosis in MRP3-positive GBM cells, demonstrated promising target antigen-restricted pro-apoptotic activity, providing a potential therapeutic approach for GBM.[197]
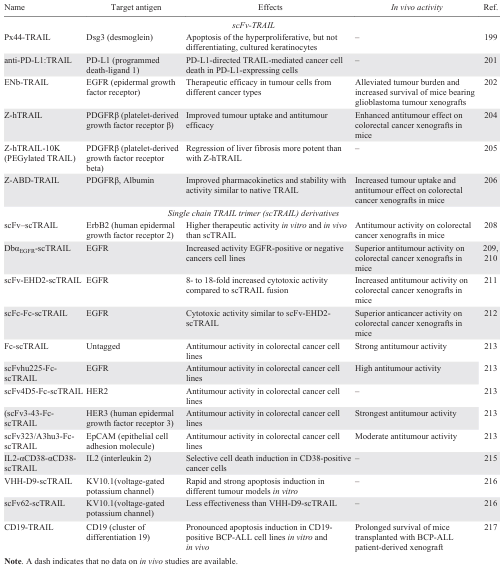
Desmoglein-3 (DSG3) has been characterized as a key mediator involved in desmosome remodelling, epidermal proliferation and differentiation, cell migration, and apoptosis, indicating that DSG3 plays a major role in tissue integrity and homeostasis. Depletion of DSG3 in HaCaT keratinocytes resulted in suppression of cell proliferation and colony growth.[198] Px44-TRAIL fusion was constructed using the anti-desmoglein mAb Px44 to target keratinocytes which express DSG3 on their surface. This fusion was biologically active and stable, and was successfully delivered to keratinocytes.[199]
Another antitumour antibody fragment with specificity for PD-L1 (programmed death ligand 1) was used to generate the anti-PD-L1:TRAIL fusion protein. PD-L1 is a key immune regulator in cancer immunotherapy, and antibodies against PD-L1 or its receptor PD-1 prevent PD-L1-mediated inhibition of antitumour T cells, thereby promoting antitumour immunity.[200] In experiments with mixed T cells and cancer cell cultures, anti-PD-L1:TRAIL fusion increased T cell activation and IFNγ secretion, resulting in increased destruction of cancer cells of various origins, including primary patient-derived cancer cells. Interestingly, anti-PD-L1:TRAIL converted immunosuppressive PD-L1-expressing myeloid cells into proapoptotic effector cells that triggered TRAIL-mediated cancer cell death.[201]
A bifunctional ENb-TRAIL fusion of the EGFR-targeting nanobody ENb derived from heavy chain-only antibodies found in camelids, and TRAIL ligand showed therapeutic efficacy in different cancer cell types unresponsive to either EGFR antagonist or TRAIL DRs agonist monotherapies. In a mouse model of primary glioblastoma, engineered stem cells (SCs) expressing ENb-TRAIL encapsulated in a synthetic extracellular matrix (SCENb-TRAIL) alleviated tumour burden and significantly prolonged survival.[202]
Platelet-derived growth factor receptor beta (PDGFRβ) is a receptor protein that is often overexpressed in tumour-associated pericytes, suggesting its potential as a target for tumour therapy. When PDGFRs are inhibited, cancer growth, metastasis, invasion and angiogenesis are reduced, enhancing the antitumour effects of cancer treatments.[203] TRAIL fusion with the anti-PDGFRβ affibody Z-hTRAIL mediated PDGFRβ-dependent binding of hTRAIL to pericytes and killed tumour cells through juxtatropic activity, or exhibited cytotoxicity in tumour cells after release from pericytes. Fusion with the anti-ZPDGFRβ affibody increased tumour uptake of hTRAIL, thereby enhancing the antitumour effect of hTRAIL in mice bearing colon carcinoma tumour xenografts without increasing acute liver and kidney toxicity.[204] Compared to hTRAIL, Z-hTRAIL showed greater in vitro cell binding and apoptosis induction in aHSCs (hepatic stellate cells). In vivo experiments showed that the anti-hepatofibrotic effect of hTRAIL was improved by PDGFRβ-targeted delivery.[205] PEGylation of Z-hTRAIL with 10 kDa PEG (polyethylene glycol) improved its pharmacokinetics and was more effective than Z-hTRAIL in resolving liver fibrosis. To improve the tumour-targeting ability and pharmacokinetics of TRAIL, a tridomain Z-ABD-TRAIL fusion protein was developed by fusing the tumour-homing ZPDGFRβ affibody and an albumin-binding domain (ABD) to the N-terminus of TRAIL. The tridomain TRAIL variant exhibited increased tumour uptake and antitumour effect in mice bearing COLO205 tumour xenografts.[206]
Members of the epidermal growth factor receptor (HER), EGFR (ERB1) and four closely related receptor tyrosine kinases receptors HER2 (ErbB2), HER3 (ErbB3), and HER4 (ErbB4) are overexpressed in various human cancers, exhibiting tyrosine kinase-dependent oncogenic activity. Numerous tyrosine kinase inhibitors and monoclonal antibodies have been approved for clinical use.[207] EGFR is both a major oncogenic driver and a therapeutic target, however current EGFR inhibitors cause cancer resistance and this remains an important unmet clinical problem. To enhance apoptotic activity, several tumour-targeting scTRAIL fusion proteins with scFv fragments specific to various HERs have been developed. ErbB2-targeting fusion scFv-scTRAIL was found to exhibit superior efficacy both in vitro and in vivo compared to non-targeted TRAIL, highlighting its potential as a promising strategy for ErbB2-targeted cancer treatment.[208]
A humanized scFv huC225 derived from cetuximab was fused to the N-terminus of scTRAIL, forming a scFvαEGFR-scTRAIL fusion, with dimerization achieved by reducing the linker between VH and VL to form a DbαEGFR-scTRAIL fusion.[209, 210] Cancer cell lines required sensitization with either cycloheximide or bortezomib to elicit efficient TRAIL-mediated induction of apoptosis in vitro by these fusions, and dimerization of the fusion enhanced the induction of apoptosis. However, in the in vivo tumour model, DbαEGFR-scTRAIL demonstrated potent antitumour activity even in the absence of chemotherapeutic agents. In addition, the dimeric tetravalent fusion protein scFv-EHD2-scTRAIL has been developed by fusing the humanized anti-EGFR scFv to the N-terminus of dimeric EHD2-scTRAIL fusion containing IgE heavy chain domain 2 (EHD2).[211] The scFv-EHD2-scTRAIL fusion was more effective in killing tumour cell and showed higher antitumour activity in murine xenograft models than EHD2-scTRAIL. Later, Fc-scTRAIL and scFc-Fc-scTRAIL fusions with enhanced antitumour activity were developed by replacing EHD2 IgGE with human IgG1 Fc.[212] Based on the Fc-scTRAIL, novel fusions were generated with scFv fragments specific for the HER2 (scFv4D5-Fc-scTRAIL) and HER3 (scFv3M6-FcscTRAIL and scFv323/A3hu3-Fc-scTRAIL) receptors, as well as the scFv323/A3hu3-Fc-scTRAIL fusion with scFv to EpCAM (epithelial cell adhesion molecule).[213] All fusions demonstrated the comparable apoptotic activity in colorectal cancer cell lines. However, in xenograft tumour model, strongest antitumour activity was observed for the anti-HER3 scFv-Fc-scTRAIL fusion with complete tumour remissions. At the same time, untagged fusion Fc-scTRAIL showed a similar in vivo activity.
CD38 has been identified as a potential target for therapeutic antibodies in the treatment of multiple myeloma, with antibody products such as daratumumab showing encouraging results in clinical trials both alone and in combination with various chemotherapies, suggesting its efficacy as a viable treatment option.[214] A novel dual cytokine–antibody fusion IL2-αCD38-αCD38-scTRAIL, designed to target CD38-positive multiple myeloma (MM) cells, exhibited exceptional biochemical properties with retained binding specificity to CD38 and a potent ability to induce selective cell death in vitro.[215]
To address the limitations of previous treatments, two fusions, scFv62-scTRAIL and VHH-D9-scTRAIL, with specificity to the voltage-gated potassium channel Kv10.1 based on the previously designed construct [184] were described. Fusion with newly generated VHH-D9 nanobody demonstrated higher antigen affinity and rapid and potent induction of apoptosis in various cell culture tumour models.[216]
A novel Fc-engineered CD19-targeting IgG1 antibody fused to a single-chain TRAIL domain, CD19-TRAIL, was developed as a new potential immunotherapeutic agent against acute lymphoblastic leukemia (BCP-ALL). This fusion demonstrated selective binding capacity and strong induction of apoptosis in CD19-positive BCP-ALL cell lines in vitro and in vivo, and significantly prolonged the survival of mice transplanted with BCP-ALL patient-derived xenografts with different cytogenetic backgrounds, especially when combined with the BCL-2 inhibitor venetoclax.[217]
A summary of TRAIL fusions with antibody fragments targeting tumour-specific antigens is presented in Table 4.
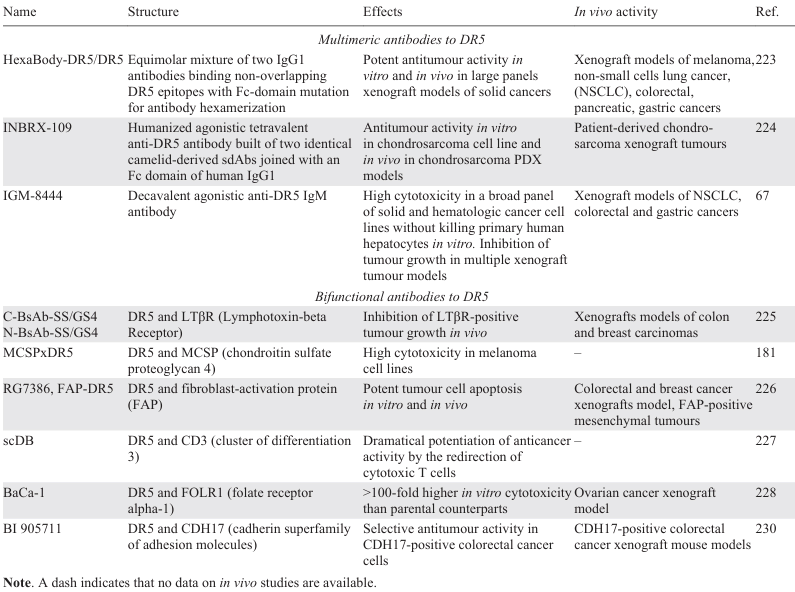
8. Multimeric and bifunctional antibodies targeting TRAIL death receptors
Over the past two decades, a variety of DR4 and DR5 receptor agonistic monoclonal antibodies have been developed. However, clinical trials of single TRAIL DR agonists or their combinations with chemotherapeutic agents have been largely disappointing.[6] In murine models in vivo, the efficacy of DR5 agonistic antibodies (such as drositumab and conatumumab) appeared to be largely dependent on FcγR-mediated cross-linking.[218-220] Therefore, novel modified versions of second-generation DR agonistic antibodies have been developed, focusing on both DR cross-linking to improve efficacy, and reducing the risk of toxicity due to non-tumour targeting effects [21].
Among multimeric antibodies, one of the first was HexaBody-DR5/DR5, an equimolar mixture of two DR5-specific IgG1 antibodies with an Fc-domain mutation that independently enhances antibody hexamerization upon FcγR-mediated crosslinking. HexaBody-DR5/DR5 has been shown to have potent antitumour activity in vitro and in vivo in large panels of patient-derived xenograft models of solid cancers.[221] It was also cytotoxic to primary cells derived from bone marrow samples of multiple myeloma patients.[222]
Another example is INBRX-109, a humanized agonistic tetravalent anti-DR5 antibody constructed of two identical camelid-derived sdAbs (heavy chain–only binding domains) linked by a on human immunoglobulin G1 (IgG1)-based Fc domain. The tetravalent format was claimed to provide a balance between high DR5 clustering efficacy and the avoidance of a potential risk of hepatotoxicity.[223] However, the decavalent agonistic IgM antibody with ten DR5 binding sites, IGM-8444, lacked the hepatotoxicity in vitro and was highly cytotoxic in a broad panel of tumour cell lines through DR5 multimerization. IGM-8444 also inhibited tumour growth in several xenograft tumour models both as monotherapy and in combination with the BCL-2 inhibitor ABT-199, and was particularly effective in a gastric PDX model.[67] All three of the above-mentioned multimeric DR5 agonists subsequently entered the clinical trials listed below in Section 9.
Alternatively, to further improve the therapeutic efficacy of anti-DR5 antibodies, several bifunctional antibodies with DR5 specificity of at least one of the moieties have been developed. Two bispecific antibodies, C-BsAb-SS/GS4 and N-BsAb-SS/GS4, were generated by fusing the anti-LTβR (lymphotoxin-β receptor) single-chain Fv (scFv) to either N- or C-terminus of the heavy chain of the anti-DR5 antibody 14A2. Both variants inhibited tumour growth of LTβR-expressing cells in vivo.[224]
The MCSPxDR5 bispecific tetravalent antibody was engineered by covalently linking a high affinity MCSP (melanoma-associated chondroitin sulfate proteoglycan) mAb complemented with a human IgG1 Fc domain to the variable binding domains of the DR5 agonistic antibody tigatuzumab for the treatment of melanoma. MCSP (CSPG4) is a promising target for cancer therapy as it is overexpressed in several malignancies and is involved in tumour growth, survival and metastasis.[180] MCSPxDR5 exhibited high affinity for MCSP, resulting in the activation of DR5 in melanoma cells. Antitumour activity of MCSPxDR5 was enhanced by FcγR-mediated cross-linking by myeloid immune effector cells.[224]
The engineered FAP-DR5 tetravalent antibody RG7386 is a bispecific antibody that simultaneously targets both fibroblast-activation protein (FAP) on cancer-associated fibroblasts in the tumour stroma and DR5 on tumour cells. FAP-driven binding of RG7386 mediated the high level of DR5 clustering required to trigger cell death. Antitumour efficacy of RG7386 was strongly FAP-dependent, and independent of FcγR-crosslinking. Treatment with RG7386 resulted in tumour cell apoptosis in vitro and significant tumour regression in several xenograft models, including patient-derived xenograft models with FAP expression either on the stroma or on malignant cells.[225]
Another research group [226] created a humanized scDB, the bispecific antibody targeting DR5 and CD3 with very low aggregate content and high stability and functionality. scDB triggered DR5 activation, and importantly, its anticancer activity was dramatically enhanced by redirecting cytotoxic T cells against both sensitive and resistant melanoma cells.
BaCa (bispecific-anchored cytotoxicity activator) is a single-agent bispecific antibody that simultaneously targets folic acid receptor alpha 1 (FOLR1) and DR5 on FOLR1-expressing ovarian cancer cells. Detailed studies showed that BaCa antibodies derived from AMG-655, lexatumumab or anti-murine DR5 antibody MD5-1 were 100 times more effective in inducing cytotoxicity in vitro than their parental counterparts. The authors hypothesized that the stronger antitumour response in vitro or in vivo may be due to BaCa supporting FcgRIIB cross-linking (Fig. 6).[227]
![[{"id":"WdIICHTER2","type":"paragraph","data":{"text":"Working model of BaCa antibody. (<i>a</i>) Healthy tissues are generally non-responsive to agonist DR5 therapy because they express no or very low level of FOLR1, thus DR5 oligomerization and activation is minimal. (<i>b</i>) In heterogeneous FOLR1-expressing OvCa cells <i>in vitro</i>, FOLR1 acts as an anchoring ligand to recruit BaCa antibody close to the DR5 antigen at the cell surface in an avidity-optimized manner. This induces a high level of DR5 clustering and activation of the apoptotic pathway in both <i>cis</i> and <i>trans</i> manner selectively in FOLR1 + OvCa cells. (<i>c</i>) <i>In vivo</i>, tumour-associated leukocytes (TAL) express inhibitory FcγRIIB receptor, which is required for the activity of DR5 agonist antibodies. Once engaged via FcγRIIB, the BaCa antibody also crosslinks the initial ternary complex (FcγRIIB-BaCa-DR5) via a FOLR1 anchor into a high-affinity stable quaternary complex (FOLR1-FcγRIIB-BaCa-DR5), which not only retains the antibody in the tumour tissue but also induces a highly superior TRAIL-R2 activation. Reproduced from Shivange et at.[[ type=\"anchor\" referenceId=\"14061\" ]] with the permission from Elsevier."}}]](/storage/images/resized/B9bS8DjhngG2uV7ZP66j2dPnEZXWkUK8KqxUnzEH_xl.webp)
Based on the anti-DR5 antibody TAS266 [228], an optimized bispecific antibody BI 905711(CDH17:TRAILR2) [229] which binds to CDH17 (cadherin-17 from the сadherin superfamily of adhesion molecules) and DR5 receptor, was created to reduce the drug-induced liver toxicity of this DR agonist. The membrane cell adhesion protein CDH17 is predominantly expressed in intestinal epithelial cells and is thought to regulate the direction and efficiency of epithelial water transport through trans-interactions with cadherins of neighboring cells.[230] It was found that BI 905711 potently triggered the extrinsic apoptosis pathway and was highly effective in several CDH17-positive colorectal xenograft models.[229] The clinical trials of BI 905711 as well as the aforementioned FAP-DR5 tetravalent antibody RG7386 are discussed below in Section 9. A summary of multimeric and bifunctional antibodies targeting DR5 receptors is presented in Table 6.
9. Clinical trials of next generation TRAIL death receptor agonists
Among the numerous newly developed next-generation TRAIL death receptor agonists, only a few have completed or are in clinical trials (Table 7). In November 2023, Aponermin (circularly permuted TRAIL, CPT) developed by Sunbio Biotech Co. Ltd. (China) received its first approval in combination with thalidomide and dexamethasone for the treatment of patients with relapsed or refractory multiple myeloma who have received at least two prior therapies.[231] This can be considered a milestone as Aponermin is the first therapy targeting TRAIL death receptors to receive clinical approval, thereby validating them as a clinically relevant target.
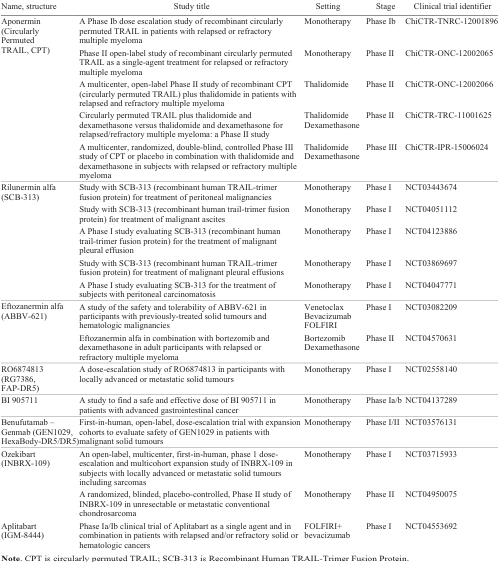
Another stabilized TRAIL variant SCB-313 (drug name Rilunermin alfa) developed by Clover Biopharmaceuticals, Ltd. (Shanghai, China) for the treatment of intracavitary malignancies including malignant ascites, malignant pleural effusions and peritoneal carcinomatosis, has completed Phase I clinical trials.[232]
ABBV-621, Eftozanermin alfa, is a hexameric TRAIL death receptor agonist being developed by AbbVie Inc. (North Chicago, Illinois USA) for the treatment of hematological malignancies and multiple solid tumours in combination with chemotherapeutic agents such as bortezomib and dexamethasone.[116] In Phase I trials, Eftosanermin alfa demonstrated acceptable tolerability and safety at a dose of 7.5 mg kg–1 once weekly, while also providing increased tumour regression in colorectal cancer.[233]
The bispecific antibody RO6874813 (RG7386, FAP-DR5) [116] has been clinically developed by the pharmaceutical company F. Hoffmann-La Roche AG (Basel, Switzerland). In a Phase I dose-escalation study, RO6874813 demonstrated a favourable safety profile in patients with various types of solid tumours, and preliminary antitumour activity was observed in patients with heavily treated NSCLC.[234]
Recently, the pharmaceutical company Boehringer Ingelheim (Ingelheim am Rhein, Germany) developed and reported the results of an open-label dose-escalation Phase Ia/b study of the bispecific antibody BI 905711 (CDH17:TRAILR2) targeting the cadherin superfamily of adhesion molecules CDH17 and the DR5 receptor (Ref. 230) in patients with advanced gastrointestinal cancer. In pre-treated patients, BI 905711 was associated with an acceptable safety profile and early signs of disease control. It is planned to continue Phase Ib clinical trials with BI 905711 using different dosing regimens (0.6/1.2/2.4 mg kg–1 every 14 days, and 0.6 mg kg–1 weekly).[235]
Among the multimeric DR5 agonistic antibodies, GEN1029 (HexaBody-DR5/DR5), named Benufutamab, was launched in 2018 in a Phase I/II clinical trial (NCT03576131) in a mixed population of patients with certain solid tumours, with support from the biotech company Genmab. However, the trial was stopped due to a narrow therapeutic window after the dose-escalation part, as explained by the sponsor.
INBRX-109 (Ozekibart) developed by Inhibrx Biosciences, Inc. (La Jolla, CA, USA) showed significant antitumour activity and a good safety profile in a Phase I trial in patients with unresectable/metastatic chondrosarcoma.[223] A randomized, placebo-controlled, Phase II trial in chondrosarcoma is currently ongoing. Importantly, the trial has received orphan drug designation from both the FDA and EMA (European Medicines Agency).
IGM Biosciences, Inc. (Mountain View, CA, USA) is currently enrolling patients in a Phase Ia/Ib clinical trial of IGM-8444 (Aplitabart) [67] as a single agent and in combination in patients with relapsed and/or refractory solid or hematologic cancers, including colorectal cancer, sarcoma, non-Hodgkin’s lymphoma, AML and chronic lymphocytic leukemia.
Finally, a PEGylated recombinant human TRAIL TLY012,[91-93] a lead product candidate from Theraly Fibrosis, Inc. (Baltimore, MD, USA), a subsidiary of D&D Pharmatech (Seongnam, South Corea), stands apart from cancer treatment. TLY012 has been granted an Orphan Drug Designation for the treatment of chronic pancreatitis in 2019 and systemic sclerosis in 2020. TLY012 is also being investigated for the treatment of non-alcoholic steatohepatitis (NASH) liver fibrosis. This confirms the potential for expanding the spectrum of application of TRAIL receptor agonists to be used not only in the treatment of cancer, but also in other diseases.
10. Conclusion
Clinical evaluation of the first-generation-TRAIL death receptor agonists has failed to demonstrate sufficient efficacy. However, the development of the novel modified versions of next-generation TRAIL death receptor agonists with enhanced antitumour properties provides confidence that targeting these receptors will eventually be brought to clinical application. A better understanding of the mechanisms by which resistance to TRAIL-mediated apoptosis signalling develops has led to the creation of new modified proteins or bispecific fusions based on death receptor agonists with potent antitumour properties. Whether next-generation drugs targeting DR4 and DR5 will be approved for cancer therapy depends on the results of preclinical or clinical trials. Particularly promising are bifunctional proteins that simultaneously target tumour cells and other components of the tumour microenvironment (immune cells, the network of tumour vasculature and other non-cellular components) surrounding the tumour are particularly promising, since it is now well established that tumour cell proliferation is largely regulated by components of the microenvironment. Optimization of bispecific protein design, such as diversification of linker-based adapters and selection of effector molecules, opens up new opportunities to increase the therapeutic index of TRAIL DR agonists.
The authors declare no conflicts of interest.
Author contributions
Alina A. Isakova, Marine E. Gasparian: writing — original draft;
Artem A. Artykov: visualization;
Dmitry A. Dolgikh: project administration, funding acquisition;
Mikhail P. Kirpichnikov: supervision;
Anne V. Yagolovich: conceptualization, data curation, writing — review and editing.
The study was financially supported by the Russian Science Foundation (Grant No. 24-14-00250, https://rscf.ru/project/24-14-00250/).
11. List of abbreviations
ABD — albumin-binding domain;
ADI — arginine deiminase;
Akt — protein kinase B;
AML — acute myeloid leukemia;
AMPs — antimicrobial peptides;
ANG2 — angiopep-2;
APAF1 — apoptotic protease activating factor 1;
BaCa — bispecific-anchored cytotoxicity activator;
Bak — BCL2 antagonist/killer 1;
Bax — member of the BCL-2 gene family;
BBB — blood–brain barrier;
BCL-2 — member of the BCL-2 family of regulator proteins;
BCL-XL — B-cell lymphoma-extra large;
BCP-ALL — B-cell precursor acute lymphoblastic leukemia;
Bfl1/A1 — BCL-2-related protein A1;
BFPs — bifunctional proteins;
BH3 — domain BCL-2 homology domain;
B-NHL — B-cell non-Hodgkin’s lymphoma;
BPL — B-cell precursor acute lymphoblastic leukemia;
CD19B — lymphocyte antigen;
CD20 — B-lymphocyte antigen;
CD3 — cluster of differentiation 3;
CD33 — myeloid cell surface antigen;
CD40 — TNF receptor superfamily member 5;
CD47 — cluster of differentiation 47;
CD7 — cell surface glycoprotein;
CD70 — cluster of differentiation 70;
CDK9 — cyclin-dependent kinase 9;
c-FLIP — FLICE-like inhibitory protein;
cIAP — cellular inhibitor of apoptosis protein-1;
CLL1 — C-type lectin-like molecule-1;
CML — chronic myeloid leukemia;
CNS — central nervous system;
CPT — circularly permuted TRAIL;
CRC — colorectal carcinoma;
CSPG4 — chondroitin sulfate proteoglycan 4;
DcR — decoy receptor 1;
DD — death domain;
DIABLO — direct inhibitor of apoptosis-binding protein with LOw pI;
DISC — death-inducing signalling complex;
DLBCL — diffuse large B-cell lymphoma;
DR — death receptor;
DRD2 — dopamine D2 receptor;
DSG3 — desmoglein-3;
EGFR — epidermal growth factor receptor;
EGP2 — epithelial glycoprotein 2;
EHD2 — EH-domain–containing protein 2;
ELPs — elastin-like polypeptides;
EMA — European medicines agency;
EpCAM — epithelial cell adhesion molecule;
ERK — extracellular signal-regulated kinases;
FA1FT — avian Ad1 spineless fibre;
FADD — Fas-associated death domain;
FAP — fibroblast activation protein;
FDA — US food and drug administration;
FOLR1 — folic acid receptor alpha 1;
GBM — glioblastoma multiforme;
HA5FT — human Ad5 fibre shaft;
hCtr1 — human copper transporter 1;
HER — human epidermal growth factor receptor;
IAPs — inhibitor of apoptosis proteins;
IC50 — half-maximal inhibitory concentration;
IDH — isocitrate dehydrogenase;
IL2 — interleukin 2;
iRGD — cyclic Peptide CRGDKGPDC;
JAK2 — Janus kinase 2;
KV10.1 — voltage-gated potassium channel;
LDM — Lidamycin;
LRP1 — low density lipoprotein receptor-related protein 1;
MAPK — mitogen-activated protein kinase;
MCL1 — induced myeloid leukemia cell differentiation protein;
MCSP — melanoma chondroitin sulfate proteoglycan;
MMAE — monomethyl auristatin E;
MOMP — mitochondrial outer membrane permeabilization;
MRP3 — multidrug resistance protein 3;
NF-Κb — nuclear factor kappa-light-chain-enhancer of activated B cells;
NRP-1 — neuropilin-1;
NSCLC — non-small-cell lung cancer;
OPG — osteoprotegerin;
Par-4 — prostate apoptosis response-4;
PDGFRβ — platelet-derived growth factor receptor beta;
PDL1 — programmed death-ligand 1;
PDXs — patient-derived xenografts;
PEG — polyethylene glycol;
PI3K — phosphoinositide 3-kinases;
RBC — red blood cell;
RIPK1 — receptor-interacting serine/threonine-protein kinase 1;
RTX — rituximab;
SAC — selective for apoptosis of cancer cells;
scFv — single-chain variable fragment;
scTRAIL — single-chain TRAIL;
SiRNA — small interfering RNA;
Smac — second mitochondria-derived activator of caspase;
Src — family of non-receptor tyrosine kinases;
SRH — antiangiogenic synthetic peptide SRHTKQRHTALH;
STAT3 — signal transducer and activator of transcription 3;
TGF3L — the third disulfide loop of TGF-Α;
TMTP1 — the synthetic 5-amino acid peptide NVVRQ;
TNC — chicken tenascin-C;
TNF — vascular endothelial growth factor receptor;
TRADD — TNF receptor-associated death domain;
TRAF2 — TNF receptor-associated factor 2;
TRAIL — tumour necrosis factor-related apoptosis-inducing ligand;
TWEAK — TNF-like weak inducer of apoptosis;
VAS — vasostatin;
VEGFR — vascular endothelial growth factor receptor;
VH — variable heavy chain;
VL — variable light chain;
XIAP — X-linked inhibitor of apoptosis protein.
References






























